


众做周知,由于微生物群落的复杂性和丰富性,精确完整的分析宏基因组极为困难。比如说,成年人的肠道中有一百种以上的不同种的微生物,如何把如此之多的微生物的基因组区分开是当前宏基因组研究的一大挑战。
95%的细菌的基因组中存在着DNA甲基化修饰:平均每种细菌有三种不同的DNA甲基化修饰序列(motifs,e.g.GATC,CTGCAG,ACCGTAGC可以是4-8个碱基,成千上万的不同可能的组合)。每一个motif在一个细菌基因组中会有几百上千次出现,而且几乎100%的出现都会被甲基化修饰。
不同种类的细菌(包括DNA序列高度相似的物种(species)或者是菌株(strains))大多拥有不同组合的DNA甲基化修饰序列motifs。从某种意义上讲,这些DNA甲基化修饰序列motif就像是天然存在于细菌DNA中的表观遗传学条形码(natural epigenetic barcode)。

本文章开发了一种在SMRT测序宏基因组数据中捕获甲基化motif的方法。
论文ID
名称:Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation
译名:利用DNA甲基化进行宏基因组binning以及分析细菌宿主基因组和相关质粒
发表期刊:Nature Biotechnology
IF:7.610
发表时间:2017年
通讯作者:Gang Fang
通讯作者单位:美国西奈山伊坎医学院(ISMMS)
样本情况
细菌样本:Bacteroides ovatus ATCC 8483等8种细菌混合培养后,提取DNA,构建PacBio 10-kb文库进行SMRT测序。
小鼠肠道微生物样本:采集12周龄小鼠粪便,提取DNA,构建PacBio 10-kb文库进行SMRT测序,并且进行16S rRNA基因V4区测序。
质粒样本:采用携带有pHel3质粒的E. coli或H. pylori提取质粒DNA,构建PacBio 2-kb文库进行SMRT测序
婴儿肠道微生物样本:两位芬兰婴儿的粪便样本,提取DNA后进行SMRT测序
分析内容
1宏基因组测序中的甲基化模式
每条序列都包含有独特的DNA甲基化特征,通过motifs计算序列的DNA甲基化得分。通过IPD(inter-pulse duration)计算motif甲基化的程度。
通常利用覆盖度和序列成分来进行宏基因组的contigs的unsupervised binning。但是这些可以通过测甲基化的方式来进行补充,可以在相似的序列成分和覆盖度下获得更好的分离contigs。也可以在微生物群体宿主细菌的contigs中定位移动遗传学元件(MGE)。
通过甲基化的方式下reads水平的聚类,可以分离得到不能被组装到contigs中的低丰度物种的reads。这些甲基化和成分特征可以和丰度特征进行整合,以最大化binning的分辨率(图1)。
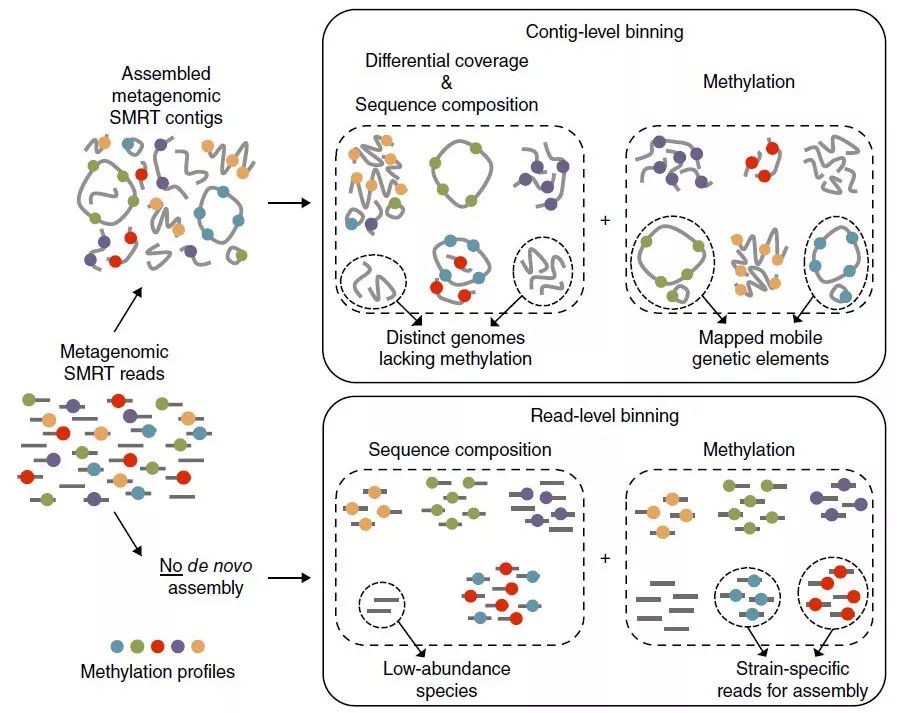
图1利用SMRT测序序列中的DNA甲基化情况进行宏基因组binning
2利用甲基化模式进行宏基因组binning
Motif甲基化得分的敏感性和特异性与IPD数量相关。每个motif的IPD的数量决定于在contig上的motif的位点数,通常短的motif的IPD比较多,另外也决定于aligning到contig的reads数量,每个read对于IPD的测量是独立影响的。(图2)
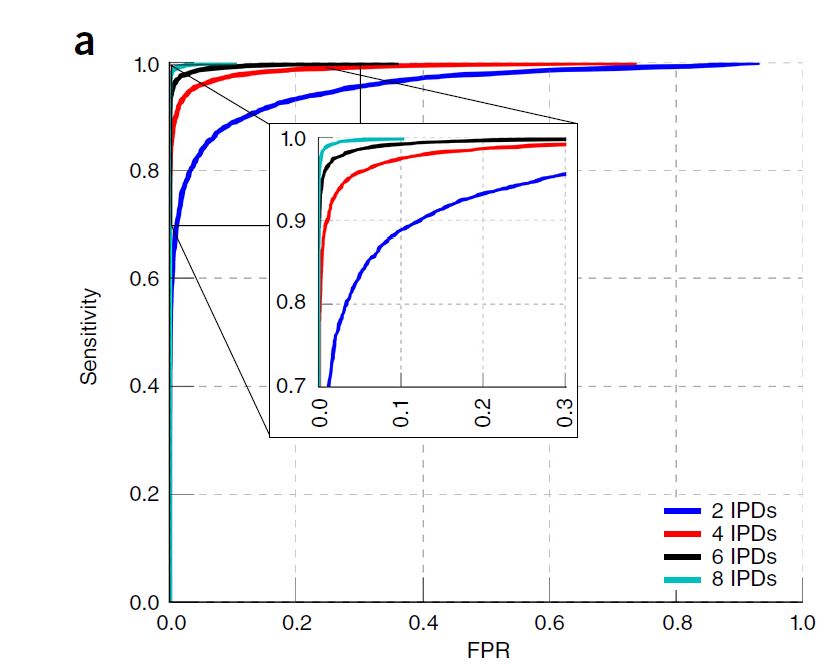
图2 ROC曲线区分序列是否甲基化
为了评估使用DNA甲基化是否可以作为宏基因组聚类的一个特征,文章首先合成了8个已经测序过的细菌的基因组,混合后利用SMRT测序。
这些混合reads通过宏基因组组装,仅基于甲基化得分,他们的motif过滤流程从宏基因组contigs中鉴别出了16个6-甲基腺嘌呤(6mA)。
其中14个(87.5%)与真正的甲基化motif准确吻合。另外2个motif:GAGC和TCACNNNNNATG与真实的motifs非常接近:GGAG and CACNNNNNATG。
对每个物种最长的contigs的motif甲基化分数分层聚类显示了每个物种在检测到的16个motif上的独特甲基化模式。(图3)
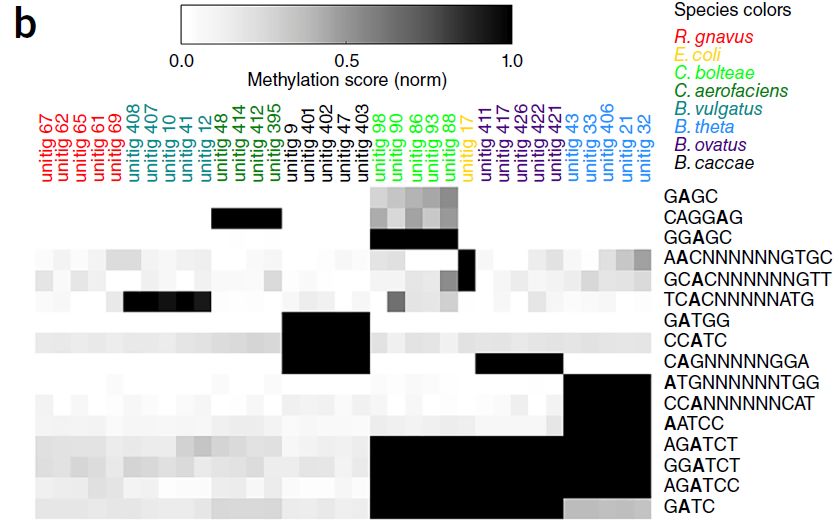
图3 Contig水平的甲基化得分热图
为了更加形象化和解释在多个宏基因组contigs上的甲基化特征,他们用了之前用于形象化展示宏基因组序列组成特征的降维规约算法t-SNE(t-distributed stochastic neighbor embedding)(图4)
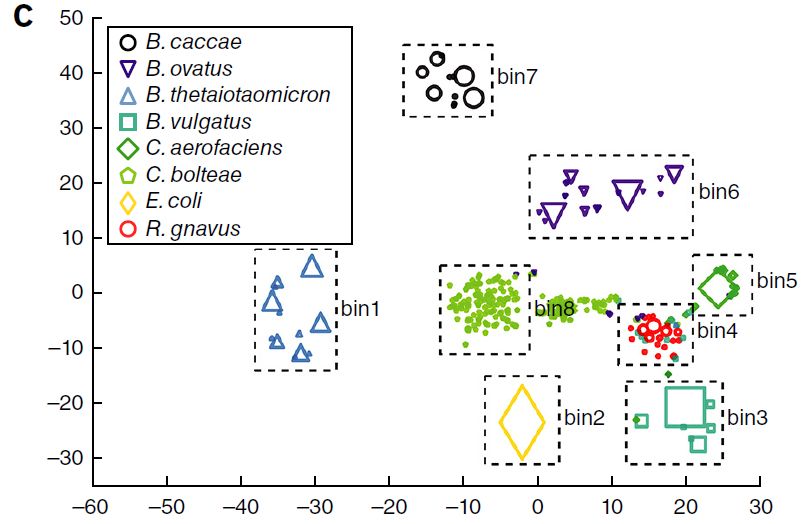
图4 16个Motif中contig水平甲基化得分t-SNE散点图
接下来他们进行了成年小鼠粪便样本的SMRT测序,分析contigs的甲基化模式。16S分析表明,这个样本的复杂度为低-中,并且有来源于拟杆菌目S24-7科的未知物种。(图5)
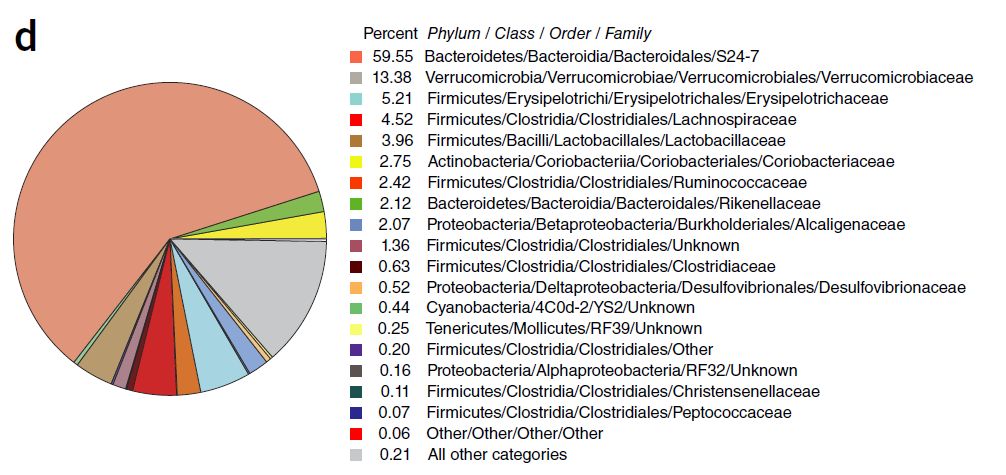
图5 成年小鼠16S科水平物种注释结果
通过motif过滤,在组装中检测到38个甲基化motifs,并且利用t-SNE方法绘图。Contigs通过Kraken进行注释。利用这38个甲基化motif的特征,鉴定到9个不同的contig bins(图6)。其中7个bins被分配到拟杆菌目,这些bins的核苷酸一致性(81-91%ANI)很高。
9个bins其中的8个,在每个bin内部align到bin contigs的reads显示一致的覆盖度水平,这表明这些bins可以对应到独立的基因组上。(图6)
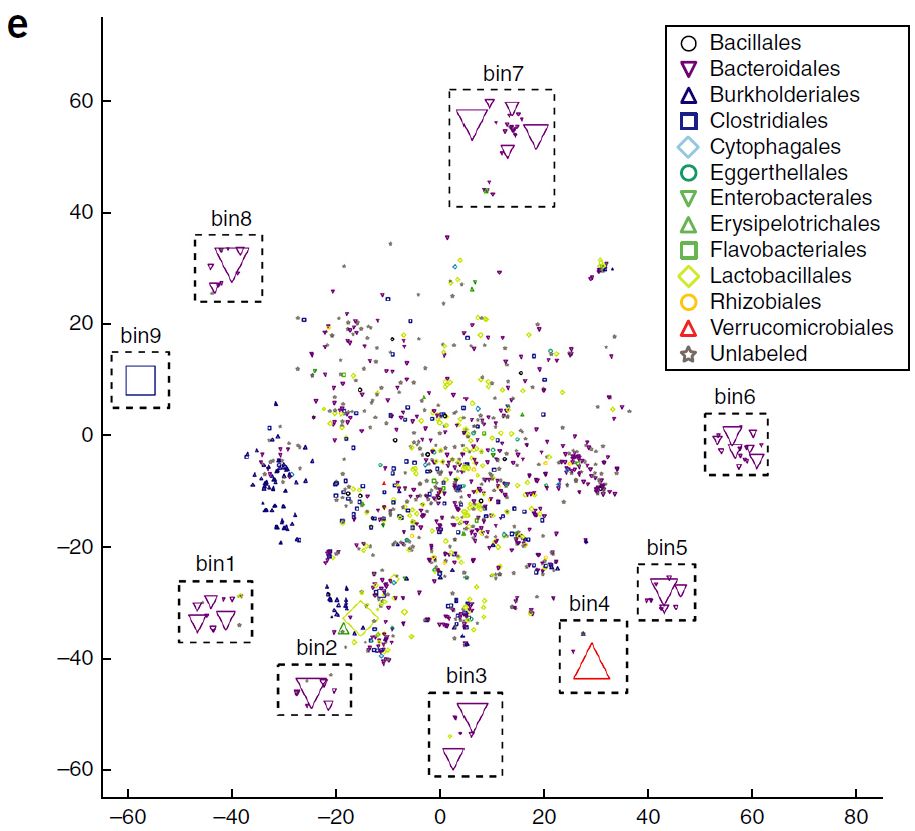
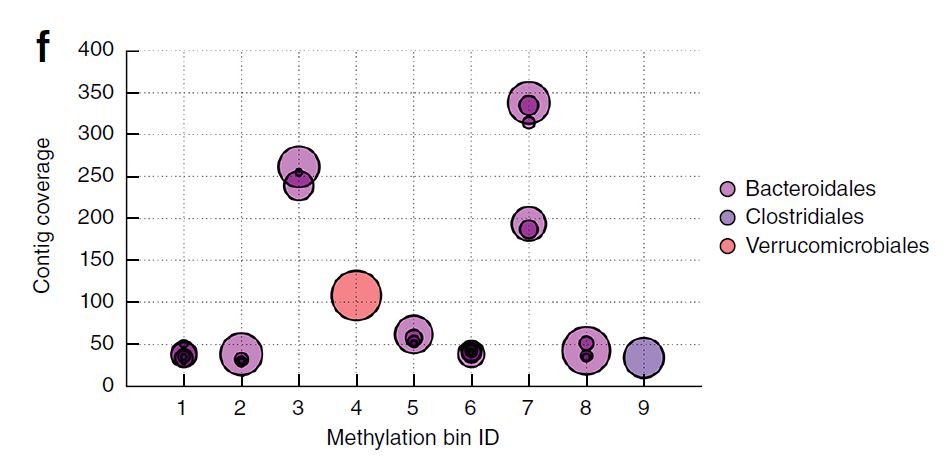
图6宏基因组contig t-SNE映射以及contig覆盖情况
3宿主染色体与可移动遗传学元件(MGEs)的关系
通过比较细菌质粒和染色体DNA的5-mer频率统计,发现其序列成分特征也会有不同,使得宏基因组样本中利用这种特征来使质粒关联宿主的分析变得不可靠。(图7)
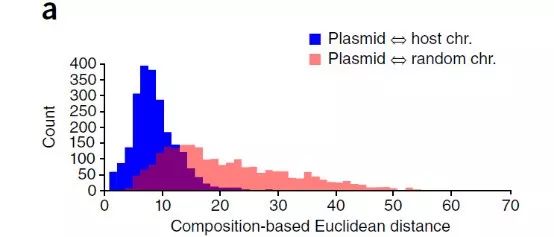
图7质粒和染色体序列的5-mer频率欧氏距离直方图
细菌的质粒和染色体DNA可以被同一套MTases所甲基化,导致甲基化特征一致。为了确认这个结果,将E.coliCFT073和Helicobacter pylori JP26转入了来源于E. coliDH5α的5.5Kb质粒pHel3。并且同时来源于这三种菌株的基因组DNA和质粒进行了测序。在每个结果中,SMRT测序结果都显示pHel3通过甲基化特征被标记了宿主菌株中。(图8)

图8 pHel3质粒和其3个宿主的甲基化模式热图
为了确定在宏基因组数据中哪种甲基化模式可以被用于在宿主基因组数据中定位质粒,首先在REBASE数据库中找出被SMRT测序的细菌染色体和质粒数据,模拟群落中有20-200个成员。在细菌群体中定位不明确的质粒需要质粒和宿主染色体基因组具有不同的甲基化组。与预期的结果一致,随着合成群落内成员的增多,独特甲基化组的数量减少,并且在同一个群落中同一个种的菌株越多的情况下,减少的越明显。(图9左)
微生物至少含有一个已知质粒的情况下,也观察到同样的趋势。(图9中)
在REBASE中已知参考序列的数据中,在随机位置提取出多个长度的核酸序列,发现平均90%的35-kb序列包含至少75%在宿主基因组中的6mA motifs,并且90%的60-kb序列捕获了100%的6mA motifs。这说明相对于小的质粒,在通过甲基化辅助binning的过程中大的质粒更有可能被正确定位。(图9右)
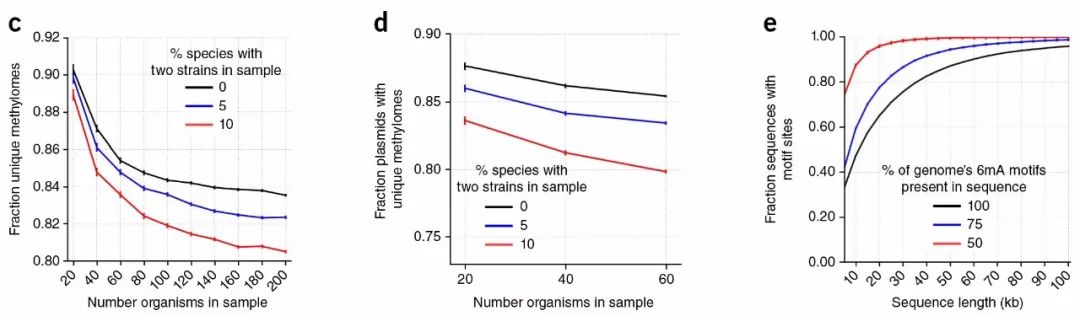
图9 SMRT测序序列的模拟分析
小 结
之前的技术很好的利用到了微生物基因组的序列信息,而最新的这项研究第一次提出微生物基因组中广泛存在、却很大程度上被忽视的表观遗传学信息(DNA甲基化修饰)可以被有效的利用于微生物群落宏基因组的研究。开启了表观遗传学在微生物群落研究中的功能性调控研究的可能性。