

皮肤局部外用仿制药研发和评价用参比制剂选择的一般考虑
Considerations on the selection of reference listed drugs for the research and evaluation of topical dermatologic generic drugs

来源

中国新药杂志, 2019 年第28 卷第6 期
作者
郭涤亮,王亚敏,马玉楠
国家药品监督管理局药品审评中心
摘要
本文参考国内外相关技术指导原则及文献,结合国内外监管机构对皮肤局部外用仿制药( TDGDs) 的评价要求,并通过阿昔洛韦乳膏的实例,分析该类药物参比制剂( RLDs) 选择的审评考虑,以期为国内仿制药企业和监管机构提供参考。
正文
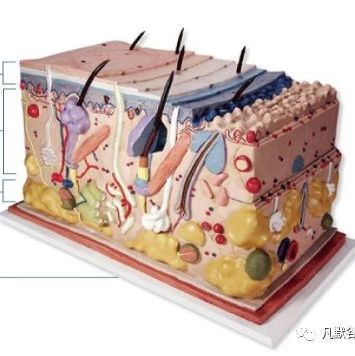
皮肤局部外用药物多为半固体剂型,如软膏、乳膏和凝胶。该类药物需要将活性成分递送到皮肤从而产生局部治疗作用,而治疗作用主要取决于递药的3 个过程:
① 活性成分从基质中释放到第一层生物屏障———角质层。
② 渗透/扩散于角质层或其他皮肤层。
③ 在作用部位产生期望的药理作用[1]。
其起效过程具有很强的变异性,从而影响药物的安全性和有效性,因此对于皮肤局部外用仿制药( TDGDs) 的开发和评价,与参比制剂( RLDs) 的质量和疗效对比研究十分关键。
RLDs 是仿制药研发和一致性评价的标杆,开展仿制药研发和一致性评价面临的首要问题就是RLDs 的选择。为规范仿制药RLDs 的选择,原国家食品药品监督管理总局( CFDA) 发布《普通口服固体制剂参比制剂选择和确定指导原则》和仿制药参比制剂目录,并建立了《中国上市药品目录集》制度。
但我国参比制剂目录及相应管理制度还处在初建阶段,并且CFDA 发布的仿制药参比制剂目录( 目前共十一批) 和《中国上市药品目录集》目前收录的品种主要为口服固体制剂,而皮肤外用仿制药研发企业在RLDs 的选择上存在诸多困惑。
本文在查阅国内外相关技术指导原则和文献的基础上,对TDGDs研究和评价用RLDs 选择的要求进行分析探讨。
1
对TDGDs 的评价考虑
国内外监管机构对TDGDs 的评价考虑不尽相同,其对TDGDs 的评价要求可以有助于分析和理解不同监管机构对该类仿制药RLDs 选择的要求和考虑。
我国尚未专门发布该类仿制药的研究和评价的指南,目前只能借鉴国外监管机构的评价要求进行研究和评价。
FDA 对于TDGDs 的评价要求也是不断明确和完善的,总体的评价考虑是在药学等效( pharmaceutical equivalents,PE) 前提下,证明其与RLDs 生物等效( bioequivalence,BE) ,通常证明BE 有体外研究和体内研究2 种方案供选择,并可采用体外研究替代体内研究。
体外研究的评价要求也日益明朗,FDA近期发布的生物等效性研究指导原则基本遵循《阿昔洛韦乳膏研究指导原则草案》[2]的评价考虑,即在保证与RLDs 处方组成( Q1) 和用量( Q2) 等同的前提下,通过特性表征、体外释放实验( in vitro release test,IVRT) 、体外透皮实验( in vitro permeation test,IVPT)并结合包装系统等对比研究最大程度地保证微观结构特性( Q3) 等同[3]。
EMA 于2015 年发布《局部外用药物质量和等效性研究指南的概念性文件》[4],在此之前也并未明确局部外用仿制药的评价要求。
该概念稿中指出对于局部外用药物在处方、剂型、生产工艺及产品使用中较小的变化就有可能对其有效性和安全性产生显著影响,通常临床试验是证明治疗等效的必要手段,虽然也存在其他手段,但大多数时候这些其他方法缺乏准确性、灵敏度或体内体外相关性。
目前,对于大多数局部外用仿制药仅与RLDs 存在PE 关系是不足以评估是否能达到治疗等效,还应进一步要求定性和定量处方组成、微观结构、物理性质、产品性能和使用等方面与RLDs 等同。
从这份概念性文件可以看出,EMA 已经意识到对于局部外用仿制药,仅保证PE 是不够的,也倾向于与FDA 类似的保持最大程度的Q1,Q2 和Q3 等同的评价考虑。
2
TDGDs 的RLDs 选择的现状
RLDs 通常是具有完整和充分的安全性、有效性研究数据作为上市依据的原研药品,因此在确立RLDs 时,其选择的首要原则为是否依据完整充分的安全性和有效性研究数据上市,其次需要关注具体品种的国内外上市背景、上市后不良反应监测情况等。
但各地区药品监管评价要求和区域内药品的上市情况等差异较大,不同地区RLDs 的确立并不统一和唯一。
各地区药品监管机构是否具有明确的TDGDs的评价要求,直接影响到各地区此类仿制药的上市药品状况和相应RLDs 的确立。以阿昔洛韦乳膏为例,中国上市的情况为进口1 家( 规格为5%) ,国内仿制39 家( 其中38 家规格为3%,1 家规格为5%) ,目前未明确局部外用药物的RLDs; 美国橙皮书查询信息显示,仅收录1 家( 规格为5%,商品名为Zovirax) ,同时标记为“RLD”和“RS”; 欧盟范围内上市约40 家,其中被认定为RLDs 的有5 个。
从以上数据可看出,对于RLDs 的选择,FDA 最为明确,其橙皮书标记为“RLD”的即为参比制剂,并且发布了《阿昔洛韦乳膏研究指导原则草案》提供了体外研究和体内研究2 种证明生物等效的研究方法供仿制药研发者选择。体外研究方法要求Q1和Q2 等同的前提下,通过特性表征、IVRT、IVPT 等对比研究最大程度地保证Q3 等同; 体内研究方法推荐为随机、双盲、平行、安慰剂对照的临床终点的生物等效性研究。
欧盟( EU) 是由多个成员国组成的联盟,各个成员国发展水平参差不齐,内部情况差异较大,并且欧盟药品上市可以选择不同的申报途径和审评程序,所以目前并没有一个统一的囊括所有已在欧盟范围内上市的药品目录以及参比制剂目录[5]。
欧盟范围内,RLDs 可选择欧盟注册目录( 通过集中审评程序上市) 、互认审评程序产品目录以及欧盟成员国产品目录( 通过成员国独立审评程序上市) 中收载的产品[6],因此存在多个RLDs 可供选择的情况,而这多个参比制剂之间是否存在PE 和BE 关系并不明确。
下面还是通过阿昔洛韦乳膏这个产品,比较欧美RLDs 选择的情况。Radulescu 和Miron[7]比对了美国橙皮书RLD( R06) ,与欧盟范围内指定为RLDs的5 个产品( R01 ~ 04 和R07) 以及欧盟市场上市的21 个仿制药( T01 ~ 21) 的处方、IVRT 及Q3 情况。
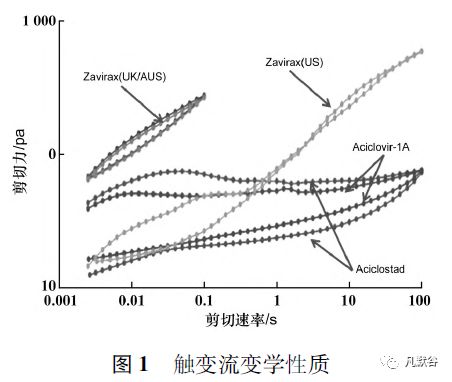
其中处方情况对比结果表明,欧盟范围内5 个RLDs( R01 ~ 04 和R07) 之间Q1 一致,但与美国橙皮书RLD( R06) 处方组成不同,主要区别在于欧盟参比制剂均采用了二甲硅油为基质,并使用了除泊洛沙姆以外的其他非离子表面活性剂( 主要为脂肪酸甘油酯类) 。
从IVRT 的单位面积药物释放量-时间平方根曲线及Q3 的动能振荡实验( Oscillatory test) 的考察结果分析,欧盟RLDs( R01 ~ 04 和R07) 与美国橙皮书RLD( R06) 差异明显,欧盟RLDs( R01 ~ 04和R07) 之间差异相对较小。
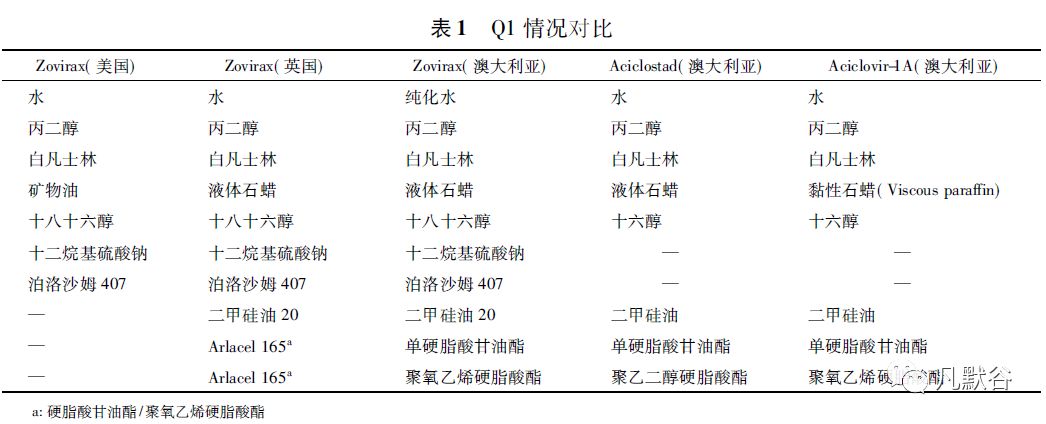
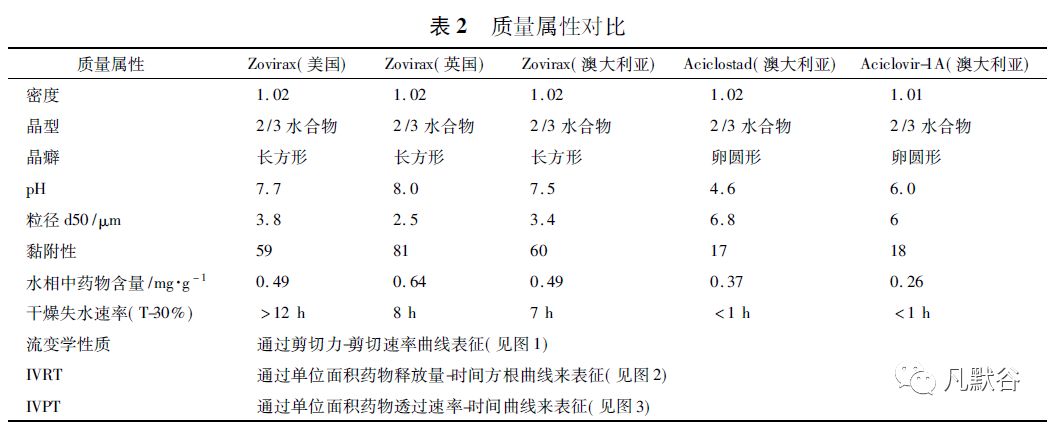
Luck[8]进一步比对在不同地区上市的原研产品Zovirax 及澳大利亚上市的2 个仿制药的Q1 情况( 见表1) 及质量属性( 见表2) ,从比对结果可以看出美国上市的Zovirax( 美国) 与欧洲、澳洲上市的Zovirax( 英国) 、Zovirax( 澳大利亚) 在处方组成上存在较大差异,与仿制药Aciclostad 和Aciclovir-1A 的差别更大; 而5 个产品的质量属性基本分为3 个差异较大的聚类:
① Zovirax( 英国) 与Zovirax( 澳大利亚) 。
② Zovirax( 美国) 。
③ Aciclostad 和Aciclovir-1A。
作者还进一步通过体内皮肤微透析试验( in vivo dermal microdialysis) 证明了Zovirax( 美国) 与Aciclovir-1A 不等效。
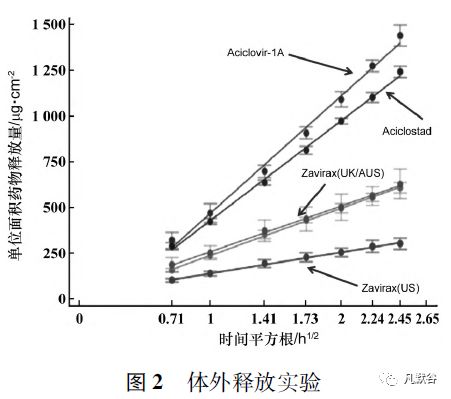
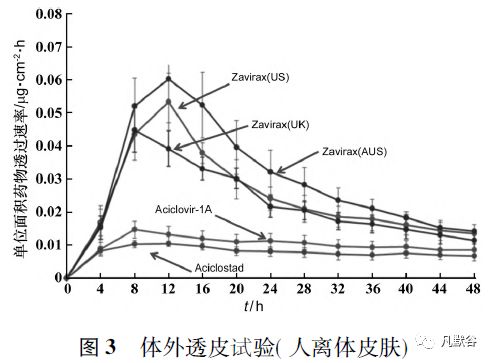
Roberts[9]通过考察不同产地原研产品的含水量,发现Zovirax( 美国) 比Zovirax( 英国) 和Zovirax( 澳大利亚) 多含10%的水,认为这可能是造成Q3差异的原因。
Trottet 等[10]通过检测139 个不同生产时期的阿昔洛韦乳膏仿制药中的丙二醇含量,发现约80% ( 139 中的111) 的仿制药中丙二醇的含量低于20%,而原研Zovirax 通过临床使用需求优化皮肤透过性确定丙二醇的含量为40%,并认为这可能是造成生物不等效的重要原因之一,例如Zovirax 的丙二醇的含量是仿制药Aciclovir-1A的2. 5 倍。
由此可见,美国与欧盟及其他地区的RLDs 或不同产地的原研产品在Q1 和Q2 存在差异,可能造成Q3 及产品安全性、有效性的差异; 而原研药品与仿制药之间,差异则更大。
因此,皮肤局部外用仿制药的研发和评价需保证RLDs 选择的准确性和唯一性,建议药品研发生产企业和监管机构可就具体产品事先确立RLDs,并建立相应的参比制剂目录。
3
讨论与总结
按我国发布的《普通口服固体制剂参比制剂选择和确定指导原则》,RLDs 首选国内上市的原研药品; 若原研药品未在国内上市或有证据证明原研药品不符合RLDs 条件,也可选用在国内上市、国际公认的同种药物( 指在欧盟、美国、日本获准上市并获得RLDs 地位的仿制药) 作为RLDs,其产品应与被列为RLDs 国家的上市药品一致; 若原研药品和国际公认的同种药物均未在国内上市,可选择在欧盟、美、日上市并被列为RLDs 的药品。
从我国现行的RLDs 选择和确定的指导原则来看,更接近世界卫生组织( WHO) 颁布的RLDs 选择标准,即要求首先以原研药品作为RLDs,对于无法确定原研药品或市场上不能获得原研药品的,可以优先选择与原研药具有同等疗效并在临床上广泛使用具有市场主导地位的仿制药或首个进入该市场的仿制药作为RLDs[11]。
但具体到皮肤外用仿制药的RLDs 选择,若原研药品未在国内上市,对于欧盟、美、日上市并被列为RLDs 的药品,也建议确定优先选择次序。
《日本医疗用医药品品质情报集》( 即日本橙皮书) 目前收载的绝大部分为口服制剂。
欧盟由于其药品上市的不同申报途径和审评程序,目前并没有一个统一的囊括所有已在欧盟范围内上市的药品目录,并且如前文分析,同一品种在欧盟范围内可能存在多个指定的参比制剂,且相互之间质量和疗效可能存在差异。
WHO 虽有较明确的RLDs 选择标准,但产品目录收载于2002 年发布的《仿制药质量等同性评价所用对照药确立指导原则》[11],品种很有限。
FDA 具有明确的TDGDs 的评价要求和RLDs 的选择规定,并建立了完善的参比制剂目录( 美国橙皮书) 。综上所述,建议优先考虑美国和WHO 确定的参比制剂,其次选择欧盟或日本指定的参比制剂,如存在多个参比制剂可供选择的情况( 如在欧盟范围内) ,建议依据具体品种的上市背景、安全性和有效性研究数据、上市后不良反应监测等情况以及多个RLDs之间的质量对比考察结果,药品研发生产企业与监管机构共同讨论确定。
参考文献
中国新药杂志, 2019 年第28 卷第6 期