
药物转运体和代谢酶在抗生素药动学中的作用
来源
药物评价研究 2017 年9 月第 40 卷第 9 期
作者
朱艳娜 ,刘克辛
大连医科大学药学院临床药理教研室
大连医科大学附属第一医院药学部
摘要
近年来,对体内药物转运体的研究取得了重大进展,越来越多的转运体被发现及研究,其对药物的跨膜转运,具有重要的意义。
各种转运体包括摄取转运体和外排转运体对药物的体内过程以及药物相互作用均有着重要影响。
研究表明大多数抗生素的体内过程都与转运体和代谢酶有关,因此,归纳总结了转运体和代谢酶在抗生素的药动学和药物相互作用中的最新研究进展,为临床合理用药提供参考。
关键词
药物转运体;抗生素;药物代谢动力学;代谢酶
正文 |
药物的体内过程包括吸收、分布、代谢和排泄,都与存在于组织生物膜上的蛋白质或多肽有关,即为药物转运体(drug transporter)。
其在药物代谢动力学以及药物相互作用中,扮演着重要角色,并影响着药物的效应和毒性[1]。
随着分子生物学技术的发展,越来越多的转运体被发现及研究,探究转运体的表达和功能,并对它们在药物体内过程中发挥的作用有了较多明确的认识;同时许多药物也被明确的证实为转运体的底物或者抑制剂,对于预测药物临床有效性和安全性有着十分重要的意义。
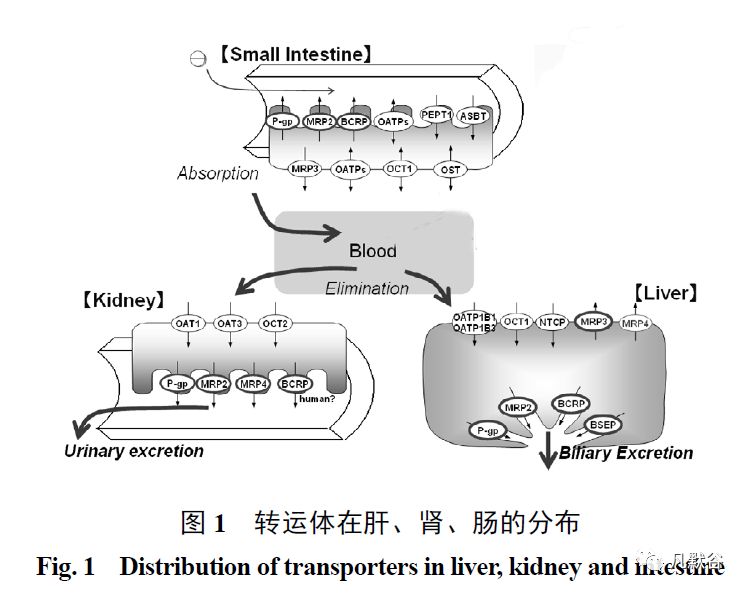
转运体在肝、肾、肠有着广泛的分布(图 1),根据底物跨膜转运方向的不同,将药物转运体分为摄取型转运体(uptake transporters)和外排型转运体(efflux transporters)。

摄取型转运体将底物摄取至靶位以发挥药效,也属于可溶性载体,包括寡肽转运体(oligopep tide transporter, PEPTs)、有机阴离子转运体(organic anion transporter, OATs)、有机阴离子转运多肽(organic anion transporting polypeptide,OATPs ) 及有机阳离子转运体( organic cationtransporter, OCTs)等。
外排型转运体是将底物逆向泵出细胞,降低底物在细胞内的浓度,属于ATP 结合转运体,包括P 糖蛋白(P-glycoprotein, P-gp)、多药耐药相关蛋白(multidrug resistance-associatedproteins, MRPs)、乳腺癌耐药蛋白(breast cancerresistance protein, BCRP ) 及多药耐药蛋白(multidrug resistance proteins, MDR)和多药及毒性化合物外排转运蛋白(mammal multidrug and toxinextrusion proteins, MATEs)等[2]。
细胞色素 P450(CYP450)是参与药物代谢的最主要的酶系统。
目前已有57 种CYP 基因在人类基因组研究中发现, 其中主要分布于肝脏的CYP1A2、CYP2C19、CYP2C9 和CYP3A4/5 参与了约80%外源药物的代谢清除[3]。
联合用药时容易发生酶所介导的药物相互作用,例如底物竞争性作用,药酶抑制或诱导作用。
抗生素在临床上应用广泛,使用率居药物之首,用来治疗和预防细菌感染。
部分抗生素通过一些摄取型转运体如OATPs 和OCTs 等进行吸收和分布[4];同时,部分抗生素通过肾脏和胆汁排泄,也需要借助于转运体的主动分泌[5]。抗生素用来控制感染,往往会联合用药来达到更好的治疗效果,而且,临床上患者病情复杂,也会多种药物同时应用,在这个过程中,就要关注由药物转运体所介导的药物相互作用。
本文归纳总结了转运体和代谢酶在抗生素的药动学和药物相互作用中的最新研究进展,为临床合理用药提供参考。
1
摄取型转运体
1.1 OATPs
OATPs 广泛的存在于各个脏器,如肝脏、肠道和血脑屏障等组织。在肝脏中主要表达的有OATP1A2、OATP1B1、OATP1B3 和OATP2B1 等,其中OATP1B1 和OATP1B3 具有特异性,在药物在肝脏消除过程中发挥着重要作用,从而影响药物的药动学过程[6]。
如他汀类降脂药物和头孢类抗生素均为其底物,被它们所转运[7,8]。
OATPs 在喹诺酮类药物的吸收和分布中也扮演着重要的角色。
非洲爪蟾卵母细胞上表达着OATP1A2,实验表明环丙沙星和左氧氟沙星可以被其转运,因此证明二者是OATP1A2 的底物[9]。
同时,另外有研究表明在大鼠中Oatp1a5 介导了环丙沙星的肠道吸收,在表达Oatp1a5 的非洲爪蟾卵母细胞中柚皮苷抑制了环丙沙星的摄取,IC50 为18μmol/L[10]。
利福平作为OATPs/Oatps 的抑制剂,当它与红霉素和克拉霉素联合应用时,OATPs/Oatps对红霉素和克拉霉素的摄取量分别降低了65%和45%[11]。
然而,利福平却没有降低这些大环内酯类药物的总血药浓度。
因此,为了了解OATPs/Oatps在大环内酯类药物的转运中是否扮演着重要角色,研究者又开始了进一步的研究。
少部分的研究表明,大环内酯类药物为OATPs/Oatps 的抑制剂。
如在OATP1B1-HEK293 细胞和OATP1B3-HEK293 细胞中,他们对典型底物如胆红素、磺溴酞(BSP)、普伐他汀的摄取量,能够被除了阿奇霉素以外的所有大环内酯类药物所抑制,并呈现浓度相关性[12]。
此外,Garver [11]的研究结果表明,在Oatp1a5-MDCK转染细胞中,阿奇霉素和克拉霉素能够显著地抑制其对牛磺胆酸盐的摄取。
相似的结果,在另一篇文献中也得到了证实,阿奇霉素和克拉霉素能够强有效地抑制Oatp1a5 对牛磺胆酸钠的摄取,Ki 值分别为3.3 μmol/L 和2.4 μmol/L。
然而,阿奇霉素和克拉霉素却不能够显著的抑制OATP2B1 对雌酮-3-硫酸酯( OATP 典型底物) 的转运。
同时, 在OATP2B1/Oatp2b1 转染细胞中,阿奇霉素和克拉霉素并没有被转运[13]。
因此,对于不同的OATPs/Oatps转运体,大环内酯类药物呈现的作用是不同的。
大部分的 β-内酰胺类抗生素都是通过载体转运而摄取进入肝脏的。
如在表达着Oatp1、Oatp2和Oatp4 的非洲爪蟾卵母细胞中,青霉素被多个转运体转运, 并且Km 值分别为4 120 mmol/L(Oatp1/Oatp1a1)、198 mmol/L(Oatp2/Oatp1a4)、1570 mmol/L(Oatp4/Oatp1b2),这个结果表明Oatp2在青霉素的肝脏摄取中占主要的作用[14]。
同时,这个研究也发现了头孢拉定、头孢唑林、头孢美唑、头孢哌酮、头孢磺啶和头孢氨苄都是Oatp2 的底物,但是头孢噻肟和头孢曲松不是其底物[14]。
另一项研究使用非洲爪蟾卵母细胞转染人的OATPs 细胞实验中,发现青霉素对于OATP1B1 和OATP1B3 的Km 值分别为74 mmol/L 和11 mmol/L。而且,其中OATP1B3 占主要作用,OATP1B1 和OATP1B3 对于青霉素在人类肝脏摄取的贡献率分别为20.5%和53.3%。
他们所检测的β-内酰胺类抗生素都能够被OATP1B3 所转运,但OATP1B1 能转运的有头孢唑啉、头孢妥仑、头孢哌酮,而不能转运头孢美唑、头孢拉定和头孢氨苄。大鼠中Oatp1a4 和人体内OATP1B3 在肝脏中青霉素摄取的作用是类似的[15]。
1.2 OATs
OATs 主要表达于近端肾小管上皮细胞基底膜侧,将血液中的有机阴离子摄取进入肾小管上皮细胞,此过程需要有机阴离子与二羧酸盐交换来提供能量[16]。
底物广泛,例如抗肿瘤药物甲氨蝶呤、乌苯美司[17];非甾体抗炎药乙酰水杨酸、吲哚美辛等。
OATs也参与了β-内酰胺类抗生素的肾脏排泄过程。如使用非洲爪蟾卵母细胞中转染表达的OAT1 细胞研究发现,头孢噻唑、头孢唑林、头孢替安和头孢氨苄都是OAT1 的底物。
在OAT3-HEK293 细胞中证实了,头孢噻啶、头孢地尼和头孢替安都是OAT3 的底物[18]。
由于OATs 的底物广泛,当两个都可以被OATs 所转运的药物联合应用时,就会发生有OATs所介导的药物相互作用。
当然,出现的相互作用也并非都是不利的。例如,当丙磺舒和青霉素联合应用之后,丙磺舒会通过抑制OATs 减少青霉素的肾脏排泄,从而提高了青霉素的血药浓度[19]。
同时,近端小管基底膜侧上的hOAT1、hOAT2、hOAT3 和刷状缘侧上表达的hOAT4 介导了四环素的摄取和外排[20]。
1.3 OCTs
OCTs 主要分布于肝和肾,其中OCT1 和OCT3主要分布于肝,OCT2 主要分布于肾。OCT 介导了重要内源性物质和药物的排泄过程。
底物广泛,临床应用的广泛的西咪替丁、二甲双胍和维拉帕米均为其底物[6]。
研究发现OCT1 主要表达在肝脏近端小管的基底膜侧,在喹诺酮类药物的代谢中起着重要的作用。
其中,环丙沙星、氟罗沙星、加替沙星、左氧氟沙星、洛美沙星、莫西沙星、氟哌酸、氧氟沙星、培氟沙星、普利沙星、米诺沙星和司帕沙星均能抑制hOCT1 对TEA 的摄取[21]。
这些药物既是OCTs 的底物又是抑制剂。
1.4 PEPTs
PEPT1 和PEPT2 表达于小肠及肾小管上皮细胞的刷状缘膜侧,调节寡肽类药物的吸收。其有广泛的底物特异性,例如β-内酰胺类药物、血管紧张素转换酶抑制剂等[22]。
PEPT1 和PEPT2 介导了头孢类药物的肠道和肾脏的重吸收,研究表明PEPT1介导了头孢拉定、头孢克肟、头孢氨苄、头孢妥仑和头孢他啶的肠道吸收[23-24];PEPT2 介导了头孢氨苄、头孢羟氨苄、头孢克洛和头孢拉定的肾脏重吸收[4, 24-26]。
它们在头孢类药物的药动学中扮演着重要的角色。
2
外排型转运体
2.1 P-gp
P-gp 是目前研究较为深入的多药耐药蛋白,它将药物从细胞内转运外排到细胞外,在体内分布极为广泛,如肠、肝、肾、脑及胎盘中均有表达[27]。
临床上极易发生有P-gp 所介导的药物相互作用,如地高辛是P-gp 的底物,抑制或增强P-gp 的转运能力会影响对其药动学过程。
Wakasugi 等[28]在研究克拉霉素对地高辛肾脏排泄的影响时,观察到地高辛血药浓度升高。
由于在地高辛的肾脏排泄过程中,P-gp 是唯一的外排转运体,推测克拉霉素作为P-gp的抑制剂,抑制了地高辛的肾排泄,使血药浓度增高。
Rengelshausen 等[29]在研究发现,地高辛和克拉霉素联合应用之后,地高辛的口服生物利用度增加,同时地高辛的肾脏排泄减少,可能是抑制了肠道和肾脏中的P-gp。
Eberl 等[30]在通过体外试验研究发现,大环内酯类药物泰利霉素、克拉霉素、罗红霉素、阿奇霉素及琥乙红霉素均可抑制P-gp 对地高辛外排转运,使地高辛血药浓度增加。
因此推测大环内酯类是有效的P-gp 抑制剂,在应用过程中要注意可能发生的P-gp 所介导的药物相互作用。
利福平作为P-gp 诱导剂,它会增加P-gp 的底物如地高辛的外排,从而降低了地高辛的血药浓度[31]。
氨基糖苷类药物大部分都是肾脏排泄,只有少部分是通过胆汁排泄。因此,外排转运体在其排泄过程中具有重要作用。
然而,目前只有一个研究发现P-gp 介导了妥布霉素的外排[32]。外排转运体对于其他氨基糖苷类药物的作用需要进行进一步的研究。
有研究表明米诺环素和利鲁唑都是P-gp 的底物,同时米诺环素还是P-gp 的抑制剂。
当二者合用时,利鲁唑在脑部的含量增加[33]。
应用Caco-2 细胞,研究者还发现土霉素也为P-gp 的底物,它可以抑制P-gp 的典型底物罗丹明123 和伊维菌素的外排[34]。
这个研究结果表明,当四环素和其他同为P-gp 底物的药物联合应用时,要注意到有P-gp 所介导的药物相互作用。
2.2 BCRP
BCRP 在体内有广泛的分布,例如小肠、肾、肝、乳腺、胰腺、胎盘以及血脑屏障等,在药物的体内过程中发挥着重要作用[22]。
能识别多种抗癌药物、膳食化合物和抗生素,其底物的专属性与P-gp和MRP2 有部分重叠。
在大鼠和人的小肠中,BCRP主要介导了环丙沙星的分泌[35]。
在另一个研究中,使用BCRP-MDCK 细胞以及Bcrp-MDCK 细胞进行实验,发现环丙沙星、氧氟沙星和诺氟沙星可以被它们所转运[36]。
环丙沙星在敲除Bcrp1 基因的小鼠中口服15 min 后的血药浓度为野生型小鼠中的2 倍[(1.77±0.73) vs (0.85±0.39)μg/mL, P<0.01]。
同时,为了检测喹诺酮类药物是否会通过Bcrp1 分泌到乳汁当中,将环丙沙星(10 mg/kg)静脉给予正在哺乳的敲除Bcrp1 基因以及野生型雌性小鼠,10 min 后收集乳汁并检测。
结果显示,在敲除Bcrp1基因小鼠中,乳汁中环丙沙星的浓度比野生型小鼠中的浓度低2 倍[ (2.19±0.13)vs(4.44±0.84)μg/mL,P<0.01]。
证实环丙沙星是通过Bcrp1 分泌到乳汁中的[36]。有趣的是,研究者还在敲除Bcrp1 基因小鼠和野生型小鼠中进行了喹诺酮类的胆汁排泄研究。
结果显示,在敲除Bcrp1 基因小鼠中喹诺酮类药物环丙沙星、格雷沙星、氧氟沙星和尤利沙星的胆汁排泄减少了,分别为野生型的86%、50%、40%和16%[37]。
因此,Bcrp1 在喹诺酮类药物胆汁排泄中具有重要作用。同时,Bcrp 还介导了头孢哌酮、头孢孟多、头孢曲松、头孢替安等头孢类药物的外排[38]。
2.3 MRPs
MRP2 主要分布于胆管、肾小管及肠道上皮细胞,MRP3 主要分布于肝血窦侧,参与胆汁排泄。
MRP2 底物广泛,包括结合型有机阴离子型化合物如内源性糖醛酸结合物、类固醇激素的葡糖醛酸及硫酸结合物、谷胱甘肽结合物以及其他外源物的Ⅱ相代谢物等的排泄[39]。
MRP2 就参与到了β-内酰胺类药物的外排过程。
例如,有研究者使用表达了rMrp2 和hMRP2 的囊泡进行试验,结果表明与空囊泡对照组相比,头孢哌酮、头孢匹胺和头孢曲松的外排量明显增加。
在只转染了hMRP2 的囊泡中,头孢替坦和头孢替安的外排相对于空囊泡也明显的增加。
上述研究表明,头孢菌素类药物是Mrp 和MRP 的底物。
Kwatra 等[40] 研究发现在MDR1-MDCKII 细胞和MRP2-MDCK 细胞中,加替沙星与红霉素联合使用后,与单独给予红霉素相比,红霉素的摄取量明显增加,分别增加了1.6 倍和1.7倍。
这提示加替沙星是一个有效的P-gp 和MRP2的抑制剂,会影响其他药物的转运
。同时,另一个实验也证实了吉米沙星是P-gp 和MRP2 的底物。
吉米沙星呈浓度相关性的抑制P-gp 和MRP2 对红霉素的外排,并且IC50 值分别为(123±2)μmol/L和(16±2)μmol/L[41]。
在Caco-2 细胞中,P-gp 抑制剂PSC833 和GF120918 以及MRP抑制剂MK571与甲磺酸达氟沙星联合应用之后,这些抑制剂可以减少甲磺酸达氟沙星的分泌从而增加其吸收率,从而证明了甲磺酸达氟沙星是P-gp 和MRP2 的底物,被其转运[42]。
上述的研究表明喹诺酮类药物是一个有效的P-gp 和MRP2 的抑制剂。
2.4 MATEs
MATEs 多表达于肝细胞胆小管面和肾小管刷状缘膜侧,将底物泵出细胞降低底物在肝及肾的浓度,对药物及其他有机阳离子的排泄过程发挥着重要作用。
对MATEs 的研究还处于初期,但已引起重视。
人MATE1 主要表达于肝脏和肾脏,MATE2和MATE2-K 主要表达于肾脏,其中MATE2-K 在肾脏有特异性表达,MATE2 在胎盘中也有表达[43]。
二甲双胍在肾脏中是通过OCT2 摄取进入肾细胞,然后再通过MATE1/2K 外排进入尿液,大概50%原型都是通过尿液排出的。
研究者为了了解抗结核药物是否会影响二甲双胍的体内过程,进行了转染细胞的摄取实验。
实验结果表明莫西沙星在OCT1、OCT3、MATE1、MATE2K 转染细胞中会显著性地抑制二甲双胍的摄取,联合用药组的摄取量仅为单独给药组的52%、39%、12%和16%。
特别是mate1、2k,抑制摄取量达到了70%以上,IC50 值分别为12μmol/L 和7.6 μmol/L。
因此,莫西沙星是MATE1和MATE2K 的有效抑制剂[44]。
3
CYP450
大 部 分 的 药物在肠道或者肝脏都要经过CYP450 代谢,CYP 酶系被抑制或者诱导,都会影响其他的药物代谢过程,也是引起药物相互作用的原因之一。
至少有7 种亚型CYP 酶(CYP1A2、CYP2C9 、CYP2C19 、CYP2D6 、CYP2E1 和CYP3A4/5)参与大量药物的氧化过程,如降血脂药、循环系统用药和抗生素类药物等[3]。
常见的抑制剂如红霉素、环丙沙星、依诺沙星、氟康唑、伊曲康唑和伏立康唑等,合并使用其他药物特别是安全范围较小的药物如华法林、环孢素、他克莫司、异烟肼等要格外注意。
4
结语
综上所述,药物转运体和代谢酶在大部分抗生素药物的体内药动学过程中扮演着重要的角色。
在临床上,患者经常会联合应用多种药物,这其中就可能包括抗生素。在这期间,要关注由药物转运体和代谢酶所介导的药物相互作用。
当两个药物共同竞争一个靶点的时候,就会发生二者的药动学和药效学的改变,甚至会引起肾脏或肝脏等组织损伤的严重不良反应。
这些研究内容将为临床联合用药提供重要依据,对促进临床合理用药具有指导意义。