
北京中医药大学
中国中医科学院中医药信息研究所
中财瀚熙(北京)生物科技发展有限公司
摘要
目的:
建立以胰脂肪酶和已上市药物为媒介的药-靶结合动力学模型,依据动力学参数与肠腔药动学构建靶点占有率模型,以评估药-靶结合动力学参数对体内药效的影响。
方法:
采用体外酶促反应体系测定胰脂肪酶抑制剂奥利司他和新利司他的半抑制浓度(IC50);通过反应进度曲线测定求解表观速率常数(kobs);采用快速稀释法测定药物-酶解离速率常数(koff);采用非线性拟合求解其他关键结合动力学参数;应用药物肠腔药代动力学模型,计算肠道中不同时间药物浓度;构建体内靶点占有率模型,并计算体内不同时间的靶点占有率。
结果:
测得奥利司他的IC50为19.1 nmol·L-1,新利司他的IC50为76 nmol·L-1;2个药物与胰脂肪酶呈时间、浓度依赖的缓慢结合反应,且结合反应属于两步结合模式,结合类型为机制B;可逆性研究结果显示奥利司他和新利司他为不可逆抑制;两药的结合速率常数(kon)分别为76.5×104和1.7×104M-1S-1,koff分别为0.33×10-6和8.4×10-6S-1。
奥利司他对脂肪酶的靶点占有率在24h大于90%,新利司他对脂肪酶的靶点占有率在21h时大于60%。
结论:
本研究所建立的基于药-靶结合动力学胰脂肪酶靶点占有率模型可用于评估动力学参数对体内药效的影响。
关键词
胰脂肪酶;药-靶结合动力学;药物肠道药动学模型;靶点占有率模型
正文 |
胰脂肪酶是一种由动物或人类胰腺分泌的水溶性脂肪分解酶,在甘油三酯的消化中发挥着重要作用,它可以消化食物中50%~70%的膳食脂肪,是人体主要的脂肪分解酶[1-2]。
随着肥胖症在全球发生率的逐年上升,药物治疗成为治疗肥胖症的有效手段之一[3-4]。
而脂肪酶抑制剂是医药界公认的相对比较安全有效的治疗肥胖的临床药物,它通过抑制胰脂肪酶的活力可有效抑制膳食中的脂肪吸收,从而达到治疗肥胖的目的[1,5]。
由瑞士罗氏公司研制的奥利司他(Orlistat,Orl)作为一种强效的胃肠道脂肪酶抑制剂,是FDA 唯一批准的减肥药[1]。
新利司他(Cetilistat,Cet)是日本武田制药有限公司研制的一种特异性胃肠道脂肪酶抑制剂[6-7]。
它们的作用机制均是通过与胰脂肪酶的丝氨酸活性位点共价结合而在小肠腔中发挥其药理活性,导致酶失活,从而阻断了人体对食物中脂肪的吸收[8-9]。
传统的药物研发采用药物-靶标结合亲和力驱动策略,认为亲和力是药理学的主要分子基础,即亲和力越强,药效就越好。
然而,最近的研究表明,与药效和药物安全性直接相关的是药物-靶标结合动力学,而非结合亲和力。
因此,药物-靶标结合动力学逐渐受到重视,药物研发策略也逐渐开始以药物结合动力学策略为主,兼顾结合亲和力[10-11]。
在这项研究中以上述2种抑制剂为工具药,以胰脂肪酶为靶点,建立了胰脂肪酶药-靶结合动力学模型;根据药物在肠道的释药动力学曲线,构建了肠道药动学预测模型;最后结合上述模型建立了药物靶点占有率预测模型。
本研究旨在通过靶点占有率模型的构建,评估基于脂肪酶靶标的结合动力学参数与体内药效的关系。
1
仪器与方法
1.1 仪器与软件
TU-1810紫外可见分光光度计(北京普析通用仪器有限责任公司);MultiSkanFC酶标仪[赛默飞世尔科技(中国)有限公司];QL-866涡旋混合器(海门市其林贝尔仪器制造有限公司);DZKW-4电热恒温水浴锅(北京中兴伟业仪器有限公司);FAI604万分之一天平(上海天平仪器厂);
JA5003B千分之一天平(上海越平仪器有限公司);SiGmA高速冷冻离心机(北京五洲东方科技发展有限公司);Brand移液枪(德国Brand公司);KQ5200E超声波清洗器(昆山市超声仪器有限公司);STARTER2100 PH 计(深圳市展华仪器仪表有限公司);GastroplusTM (Version 9.5.0004Simulations Plus Inc.,Lancaster,CA,USA);ADMET PredictorTM软件(Version 8.1.0.0)。
1.2 试剂
猪胰脂肪酶(上海源叶生物有限公司);对硝基苯基棕榈酸酯(P-NPP)(≥98%,阿法埃莎化学有限公司);脱氧胆酸钠(纯度:>99.0%,上海江莱生物科技有限公司);二甲基亚砜(DMSO)(北京鼎国昌盛生物技术有限责任公司);
磷酸二氢钠、异丙醇、高氯酸(北京化工厂)、氢氧化钠(天津市光复科技发展有限公司)均为分析纯;奥利司他(纯度:≥98%,中山万汉医药科技有限公司)、新利司他(纯度:≥98%,上海源叶生物科技有限公司);娃哈哈饮用纯净水(杭州集团娃哈哈有限公司)。
1.3 方法
1.3.1 胰脂肪酶溶液的制备
精密称定胰脂肪酶100mg,于50mL 离心管,加入10mL 冷的50mmol·L-1磷酸盐-5mmol·L-1脱氧胆酸钠缓冲液(pH8.0),3000r·min-1涡旋5min,4℃下离心半径7.4cm,7000r·min-1离心10min,倾出上清液至一新的离心管,即得。
使用时保存在0~6℃冰水浴环境。
1.3.2 胰脂肪酶抑制剂IC50的测定
精密称取奥利司他、新利司他各10mg至于10mL 量瓶中,DMSO溶解并定容,冷藏储存,使用前取出室温融化,摇匀后以缓冲液稀释得系列不同浓度的抑制剂溶液(奥利司他终浓度0~2880 nmol·L-1;新利司他终浓度0~6144 nmol·L-1)。
分别取0.3mL抑制剂溶液和上述已制备的酶溶液于试管中,加缓冲液补至2.7 mL,摇匀,37℃水浴温孵15min,加入1mmol·L-1p-NPP底物溶液0.3mL,37℃水浴10min后立即于405nm测定其吸光度,空白以等量不含抑制剂的溶液代替,每组反应平行3次。
IC50计算采用公式(1),其中K为曲线的上限值,a 为增长速度,采用三参数logistic曲线拟合求解。
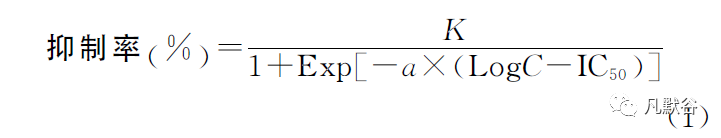
1.3.3 反应进度曲线的测定
分别精密量取20μL0.5mmol·L-1P-NPP溶液、20μL系列不同浓度的奥利司他(或新利司他)溶液,120μL缓冲液于96孔酶标板,振荡,37℃温孵10min后加入胰脂肪酶(5mg·mL-1)40μL启动反应,每隔10s读数1次。
以加入相应溶液的溶剂作空白对照,计算之前减去空白吸光度值,每组反应平行3次,最后取其均值。
根据Williams and Morrrison方程(公式2)求解kobs。
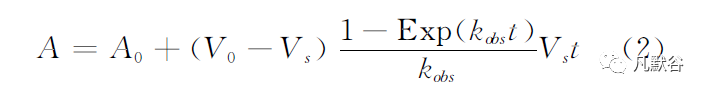
式中A 为吸光度,A0为空白吸光度,V0和Vs为起始和稳态反应速度,t为时间。
计算获取kobs后,根据方程(3)非线性拟合求解Ki(初始结合一级速率常数)和ki(缓慢反应一级速率常数)。
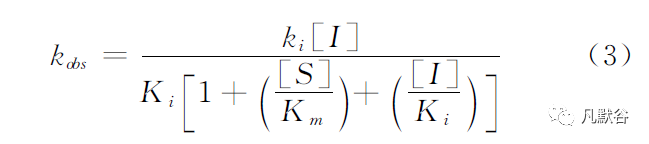
再依据方程(4)求解kon。

1.3.4 解离速率常数测定
采用快速稀释测试法[12]。
等份胰脂肪酶,一份加抑制剂,一份不加,平行对照空白酶组和空白底物组。
37℃温孵15min后用Spin富集技术,去除未与酶结合的游离抑制剂,富集抑制剂-酶复合物,然后快速10倍稀释到含有过量底物P-NPP(0.15 mmol·L-1)的缓冲液中,以最小化药物再结合对解离速率测定的影响。
通过产物生成量监测酶-抑制剂复合物的解离,每10s测定一次。
观察经去除游离抑制剂处理后酶活恢复趋势,依据方程(5)和(6)计算koff。式中Vs为未抑制酶反应速度,C 为空白背景吸光度;V0为起始反应速度,t为时间。
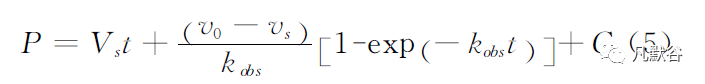

1.3.5 肠道药动学(IPK)模型的建立
收集奥利司他、新利司他模型建立参数,使用Gastroplus 9.6中的ADMET PredictosTM(Version 8.1.0.0)模块进行预测基本建模参数,药物渗透性参数采用软件进行优化获得。
模型建立中所用的药物的理化性质参数、生物药剂学参数见表1。
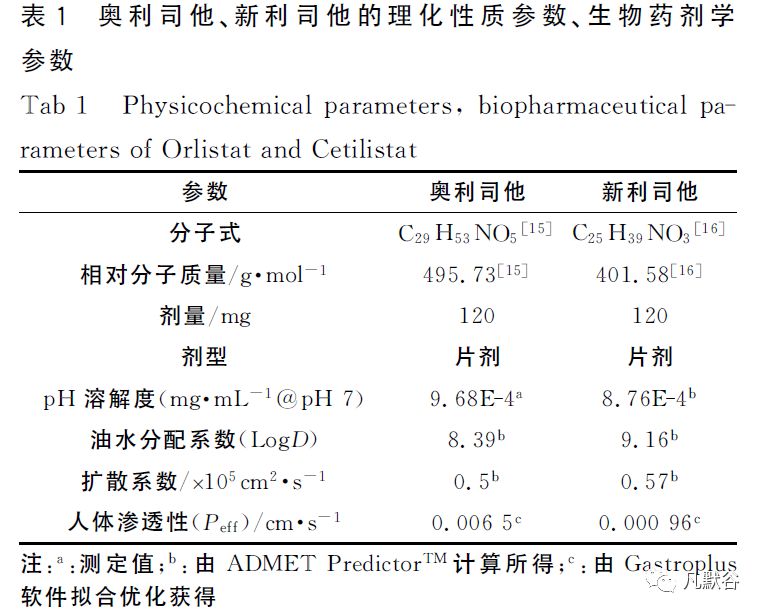
由于奥利司他和新利司他均不吸收入血(生物利用度小于1%[13-14]),因此模型构建时不考虑加载药物在体内处置过程的参数。
模型评估采用实测生物利用度数据同IPK模型预测结果进行比较验证,以评估模型建立符合要求。
采用该模型预测药物在肠道中(主要研究空肠)的不同时间药物浓度(IPK),并计算药物空肠清除半衰期(t1/2β)。
1.3.6 体内靶点占有率模型(BK-IPK模型)建立
该模型主要是采用IPK关键参数(CI:不同时间空肠肠腔药物浓度)和药物-靶标结合动力学(BK)关键参数(kon,koff)构建。
该模型药物-靶标的结合和平衡两个过程均采用BK和IPK参数来表征,解离过程采用单独BK参数(
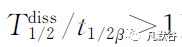
)联合表征。
模型方程如下。
其中[TC]为药物-靶标复合物浓度,[T]为靶点浓度,[CI]为肠腔药物浓度,t 为时间。
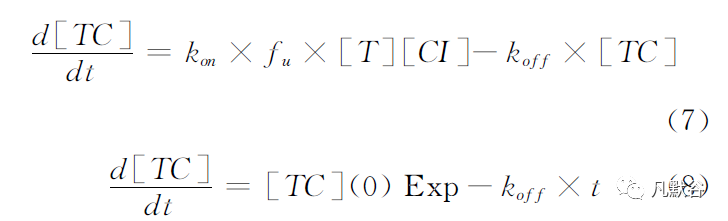
2
结果
2.1 胰脂肪酶抑制剂IC50测定
胰脂肪酶作用于P-NPP分解出对硝基苯酚(P-NP),该产物在405nm 处有吸收峰,浓度与吸光值成正比。测定奥利司他、新利司他在不同浓度条件下与酶反应后的吸光度,从而得出其对胰脂肪酶的抑制活性。
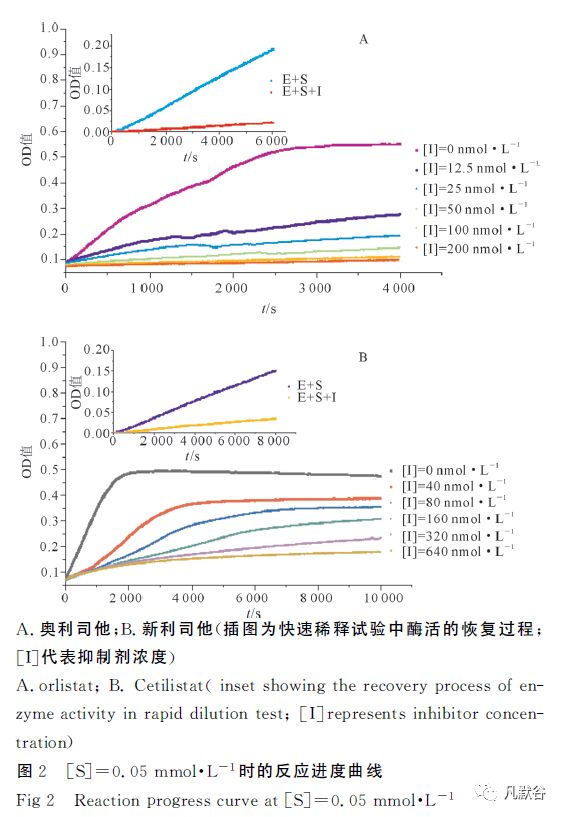
奥利司他、新利司他对胰脂肪酶的抑制考察分别如图1(A、B)所示,随着抑制剂浓度的增大,抑制率快速增大,最大抑制率均近100%。
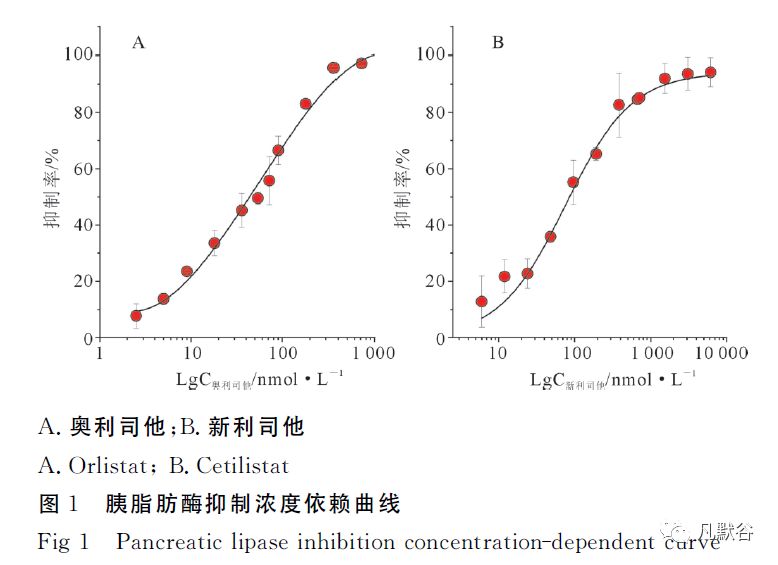
采用公式(1)计算得奥利司他的IC50为19.1 nmol·L-1,新利司他的IC50为76 nmol·L-1。
2.2 反应进度曲线测定
如图2A、B所示,奥利司他、新利司他展现出相似的抑制机制。
在不同浓度奥利司他、新利司他存在下,接近稳态时的反应进度曲线呈现出明显的时间和浓度依赖,这是缓慢结合抑制动力学的特征[17]。
从快速稀释试验中酶活的恢复过程(图2A、B插图)可以看出随着反应时间的延长酶活基本未有明显恢复或恢复极其缓慢,故2个药物均应该是胰脂肪酶的不可逆抑制剂或解离速度极慢的可逆结合抑制剂,不可逆结合可能与文献报道的2种抑制剂均为与脂肪酶的活性丝氨酸部位共价键合保持一致[8-9]。
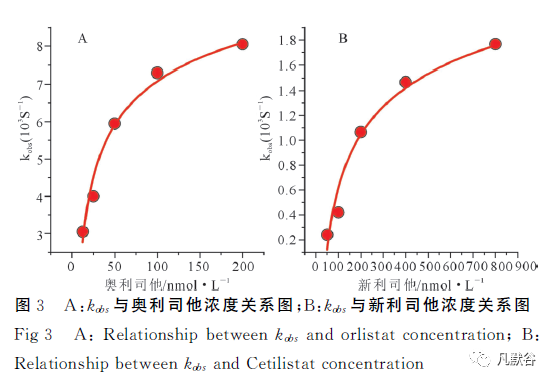
计算kobs后,将kobs和不同抑制剂浓度作图,如图3A、B所示,两者呈双曲线函数关系,kobs随抑制剂浓度的增加而呈上升趋势,这种行为是典型的缓慢结合二步诱导契合结合反应类型[18],且结合类型属于机制B,即药物与脂肪酶快速结合,不稳定的药物-酶复合物过渡态然后缓慢异构化形成稳定的复合物。
2.3 解离速率常数测定
采用Spin技术富集抑制剂-酶复合物,酶回收率>95%,抑制剂的去除率>99.5%,方法验证符合要求。
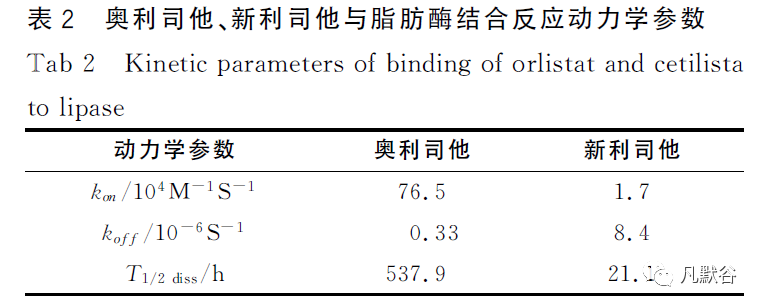
采用方程(5)拟合数据求解koff,2个抑制剂的koff值计算结果见表2。
一般该值求解可采用文献[19]报道的线性拟合方程计算,但当底物浓度不够大时,kobs与抑制剂浓度[I]线性关系不理想会导致计算koff值误差过大,实验中采用线性方法计算的结果显示奥利司他的koff值是指数方法的2000倍,新利司他计算koff值结果是负值。
本试验研究中发现底物浓度过大时,会对实验结果产生较大影响,因此不宜增加底物浓度,所以采用快速稀释法计算koff值。
从实验中求解的主要动力学参数计算结果见表2。
2.4 肠道药动学(IPK)模型的建立
采用健康男性受试者口服给药120mg后实测的生物利用度数据[13-14]与所构建的IPK 模型预测结果进行对比验证。
奥利司他和新利司他属于极低渗透性药物,基本上不吸收入血,生物利用度约为1.0%。IPK 模型计算的奥利司他和新利司他生物利用度为0.9%和0.8%,预测值和实测值的倍数误差在2倍范围内[20];
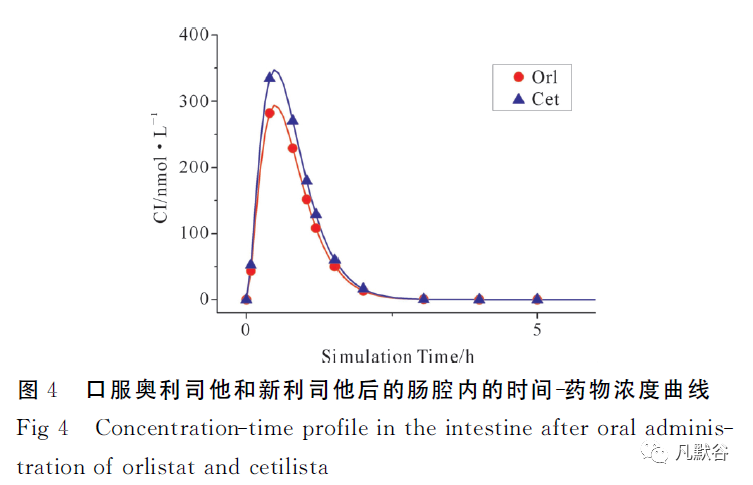
并且IPK模型计算的奥利司他和新利司他的药物吸收百分率分别为3%和5%,这也表明药物为极低渗透性,这与文献[21]报道的数据基本一致,这表明所建立的IPK 模型能够较好地模拟人体口服给予奥利司他和新利司他后的肠腔内的时间-药物浓度曲线。
IPK药-时曲线数如图4所示。
2.5 体内靶点占有率模型(BK-IPK模型)建立
采用BK 和IPK 参数数据组合评估奥利司他和新利司他对胰脂肪酶占有的结合和平衡过程的影响(此处只考虑2个药物与酶形成的复合物解离极缓慢的情况)。
奥利司他和新利司他的药-靶结合半衰期均远大于IPK半衰期,这表明模型的解离过程采用药物-标靶半衰期(
)来表征此过程是更合适的。
如图5所示,模型预测奥利司他和新利司他与脂肪酶的靶点占有率达到90%的时间分别0.08 h和0.72h,预测的最大靶点占有率分别为99.8%和93.0%,奥利司他脂肪酶的靶点占有率在24h仍大于90%,新利司他对脂肪酶的靶点占有率在21 h时大于60%。
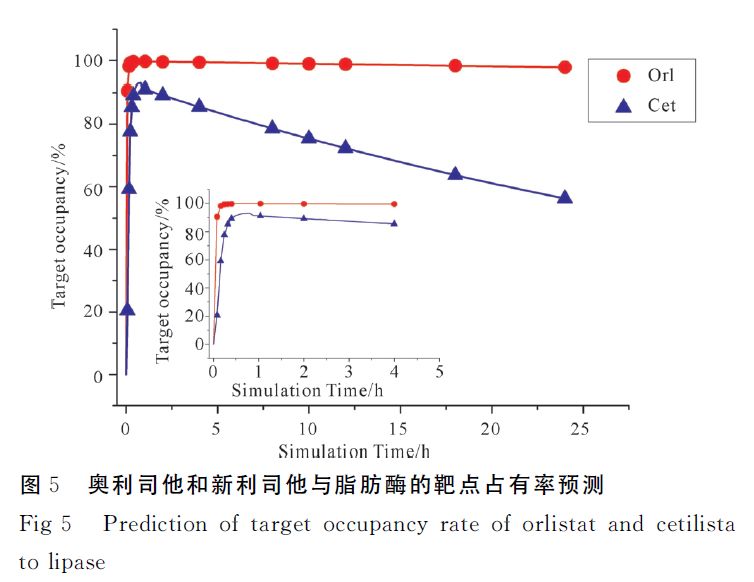
3
讨论
奥利司他和新利司他作为强效的胃肠道脂肪酶抑制剂,是目前公认的用于治疗肥胖症的药物。
本研究测定得奥利司他的IC50为19.1nmol·L-1,新利司他的IC50为76 nmol·L-1,均为纳摩尔级抑制,与文献报道值接近[22-23]。
尽管IC50对评估药物对酶的潜在抑制作用有较大的价值,但因其只能反应药物-靶标结合的平衡状态,不能反映整个结合的动力学过程,且无法预测药物对酶抑制的快慢和作用持续的时间等,存在较大局限性使其无法与体内的药效活性建立关联。
本研究构建了奥利司他和新利司他与脂肪酶的结合动力学模型,探索了两个抑制剂和酶的结合机制。
奥利司他和新利司他均属于不可逆结合抑制或属于可逆结合但复合物解离极缓慢抑制,反应速率具有时间和抑制剂浓度依赖,属于二步诱导契合结合机制。
计算两个抑制的Ki*值分别为0.43pmol·L-1和0.49 nmol·L-1,表明2个抑制剂与脂肪酶的结合过程属于缓慢紧密结合。
并在结合动力学模型基础上构建了BK-IPK 模型,将体外的药-酶动力学参数和体内药效作用建立关联,以评估kon和koff对药物体内最大靶点占有率和靶点占有时间的影响。
奥利司他的kon值更大,koff值更小,结合速率常数是新利司他的45倍。
这表明奥利司他与脂肪酶形成复合物过渡态的能量更低,与底物(脂肪)具有更强的竞争力,与酶具有更快的结合速率。
临床上研究结果表明[24-26],奥利司他制剂可以在饭后1h内服用,但新利司他制剂需要在饭后立即服用,体外kon值的结果与两药临床试验结果相一致;采用BK-IPK模型计算得二药在体内的靶点占有率均可达90%以上,因此二者服用相同剂量即可(均为口服120mg)。
奥利司他的koff值更小,药物-靶标复合物半衰期更长。
根据BK-IPK模型预测,在24h时奥利司他的靶点占有率仍大于90%,理论上奥利司他对脂肪酶的抑制可以持续更长时间,该药在体内的药效至少可持续1d,但由于机体在餐后脂肪酶会由胰脏重新分泌排到十二指肠腔中,所以两药在每次用餐时需要重新服药。
BK研究中最主要的动力学参数是koff,该值拟合计算常采用线性拟合方程求解,但一般当抑制剂和酶属于缓慢结合反应时,只有底物浓度大时,通过反应进度曲线测定的kobs和[I]才可能产生良好的线性关系,此时计算的才更准确,否则计算出koff值可能误差过大。
采用快速稀释法测定koff值时,需要控制游离抑制剂浓度远低于Ki值,一般该实验测定结果也会受到多种因素的影响,比如靶点再结合,药-酶初始结合速率等。
一般来说,测定的结果koff≥kobs值,也就是实际的结果会略高于拟合计算的结果。
但本研究中由于2个抑制剂均属于与酶结合速率较快(第一步解离速率常数较小),并且不会产生靶点再结合的情况,所以计算的数据结果准确度更高。
当前药物活性筛选的主流优化策略是减小IC50值和增加药物的血浆稳定性,而本研究结果是对主流优化的一种挑战:
(1)IC50在一定程度上,并不会影响到药物体内的靶点占有率,不必过于追求更小的IC50值;
(2)从理论上说,主靶点(产生药效)和副靶点(可能产生副作用)的BK曲线具有足够的区分度,药物具备足够优良的BK参数时,甚至体内快速的药物清除率也是我们期望的优化方向,因为这不仅不会降低体内的靶点占有率,反而会因为药物快速清除而减少产生更多的副作用。
因此,本研究为化合物活性筛选和药物设计提供了一种新的思路和方法。