
作者
田禾,马昌友,吴舰,徐丹
南京正大天晴制药有限公司
摘要
Akt,即蛋白激酶B,是磷脂酰肌醇3-激酶(PI3K)信号通路重要成员。
超过50%的肿瘤中Akt过度活化,因此对Akt的抑制也是肿瘤靶向药物开发的方向之一。
Akt发现至今已近30年,但尚无小分子抑制剂成功上市。
近期Akt抑制剂ipatasertib和capivasertib的成功研发,以及对三阴性乳腺癌和转移性去势抵抗性前列腺癌Ⅱ期临床研究取得的较好疗效,确证了Akt抑制剂对PIK3CA/AKT1/PTEN基因突变或缺失患者的治疗优势,也指明了激酶选择性更好的ATP竞争性抑制剂是未来的发展方向。
关键词
蛋白激酶B;ipatasertib;capivasertib;ATP竞争性抑制剂
正文 |
2018年美国临床肿瘤学会(ASCO)年会上,阿斯利康公司公开了旗下在研的Akt抑制剂capivasertib(AZD-5363)联用紫杉醇一线治疗转移性三阴性乳腺癌(TNBC)的临床试验结果。
在一项名为PAKT的随机、双盲、安慰剂对照的Ⅱ期临床试验中,与对照组(紫杉醇+安慰剂)比较,治疗组(capivasertib+紫杉醇)中位无进展生存期(PFS)由4.2个月提高至5.9个月,达到了主要研究终点[1]。
在对患者的亚组分析中,20%的入组患者存在PIK3CA、AKT1或PTEN基因突变或缺失,对应治疗组和安慰剂组的中位PFS分别为9.3个月和3.7个月,获益明显[2]。
此前,罗氏的Akt抑制剂ipatasertib针对TNBC的Ⅱ期临床试验(LOTUS)也得到了类似的结果[3]。
Akt作为缺乏有效靶向药物的TNBC的治疗靶点再次受到广泛关注。Akt是PI3K/Akt/mTOR信号通路重要成员,通过下游众多效应器影响细胞存活、生长、代谢、增殖、迁移和分化。
据统计,超过50%的肿瘤Akt过度活化,尤以前列腺癌、胰腺癌、膀胱癌、卵巢癌、乳腺癌为主,Akt过度活化可导致肿瘤发生、转移以及耐药性的产生[4-6]。
抑制Akt活性既可避免抑制上游PI3K造成的严重副作用,也可避免抑制下游mTOR引起的负反馈机制影响药效[7]。
因此,寻找高效和选择性好的Akt抑制剂是当前肿瘤靶向药物研发的重要方向。
本文对当前在研的Akt抑制剂临床研究进展进行介绍。
Akt结构及活化机制Akt发现至今已近30年,迄今未有靶向药物上市。
1987年,STAAL等[8]从源自AKR品系小鼠胸腺瘤(thymoma)细胞系分离得到的逆转录病毒AKT8中首次鉴定和克隆了v-akt基因,并克隆了v-akt人的同源基因AKT1和AKT2。
1991年,3个独立实验室采用不同策略分别确认了v-akt编码的产物———分子量为57000的丝氨酸/苏氨酸蛋白激酶[9-11]。BELLACOSA等[9]通过cDNA与v-akt杂交来克隆并表达蛋白激酶,因此该激酶被命名为c-Akt。
COFFER等[10]经由cDNA文库筛选,找到一个与蛋白激酶A和C极其相似的蛋白激酶,命名为蛋白激酶B(PKB)。
JONES等[11]基于简并PCR技术编码激酶结构域,命名为RAC———激酶A和激酶C相关的激酶。
这也是Akt又被称为PKB的原因,而另一名称RAC,为避免与Ras通路的GTP酶Rac混淆,目前已不再使用[12]。
Akt包括3种亚型,即Akt1、Akt2和Akt3。
作为典型的蛋白激酶,每种亚型均由氨基端的PH结构域,结合ATP的激酶结构域以及羧基端的调节结构域组成(图1)。
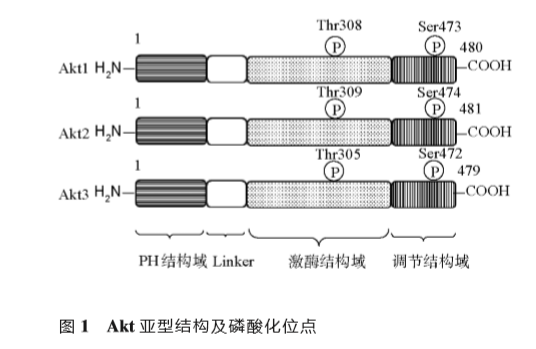
3种亚型约80%的氨基酸序列同源,仅在PH结构域和激酶结构域连接处变化较大[13]。Akt3种亚型的活化方式基本一致。
通常情况下,Akt以无活性的形式存在于细胞质中,PH结构域与激酶结构域的分子内相互作用形成“PH-in”构象,使激酶结构域活化环(activationloop)不能被3-磷酸肌醇依赖性蛋白激酶1(PDK1)磷酸化[14]。
Akt在细胞膜或内膜上完成激活,激活途径有所差别。
在细胞膜表面,激活的受体酪氨酸激酶(RTKs)或G蛋白偶联受体(GPCRs)活化PI3K,PI3K磷酸化磷脂酰肌醇4,5-双磷酸(PIP2)于细胞膜内侧生成3,4,5-三磷酸磷脂酰肌醇(PIP3),促使Akt转位至细胞膜以PH结构域结合PIP3,Akt构象改变为“PH-out”,暴露出激酶结构域磷酸化位点,使Akt1的T308(Akt2-T309,Akt3T305)磷酸化。
Akt去活化则由PTEN、PP2A、PHLPP等磷酸化酶控制。在内膜系统上,Akt的PH结构域结合磷脂酰肌醇3,4-双磷酸,由SHIP磷酸化酶激活,并由INPP4B抑制[15,16]。
Akt抑制剂Akt有众多结合位点,设计药物占据这些位点可以阻断Akt的某些功能,从而影响信号通路。
根据Akt的结构及与抑制剂结合位点的不同,可将药物分为PH结构域抑制剂、变构抑制剂和ATP竞争性抑制剂3类,结构式见图2所示。

早期的Akt抑制剂多是PH结构域抑制剂和变构抑制剂,ATP竞争性抑制剂报道较少,这是因为ATP竞争性抑制剂曾被认为缺乏激酶选择性,即靶蛋白Akt与AGC激酶家族各激酶(如PKA、PKC、p70S6K等)同源性高,ATP结合位点的结构相似,该类抑制剂同时也能抑制其他AGC激酶,进而导致脱靶毒性。
但ATP竞争性抑制剂具有仅作用于活化的Akt、不抑制非磷酸化无活性的Akt的优点,成药潜力大于PH结构域抑制剂和变构抑制剂。随着ipatasertib等高选择性ATP竞争性抑制剂的开发,当前在研Akt抑制剂以ATP竞争性抑制剂为主。
1
PH结构域抑制剂
Akt的PH结构域结合PIP3,通过抑制剂竞争性结合PH结构域,可阻止Akt向细胞膜的转位及PDK1和mTOR2的磷酸化。
根据抑制剂化学结构的不同又可分为磷酸肌醇类似物(PIA,如PIA5)、烷基磷脂(如edelfosine、miltefosine和perifosine)、肌醇多磷酸(如2-O-BnInsP5)、磺胺类(如PHT-427)、嘌呤或嘧啶类似物(如triciribine)和其他类型(如PIT-1)[17]。
虽然这些化合物表现出一定的体外抑制活性,但是理化和药动学性质较差,多处于临床前研究阶段即终止。
perifosine由AeternaZentaris开发,是最早进入临床试验的Akt抑制剂[7]。
perifosine单药治疗肾细胞癌疗效并未优于现有药物如替西罗莫司和依维莫司等[18]。
有研究报道perifosine与卡培他滨联合用于转移性结直肠癌的治疗,Ⅲ期临床试验结果显示中位总生存期(OS)为6.4个月,与单用卡培他滨(6.8个月)相比未提高(P=0.315)[19]。
另一项与硼替佐米和地塞米松联合用于治疗多发性骨髓瘤的Ⅲ期临床试验,也因中期分析难以达到PFS的显著差异而停止[20]。
目前仅有一项perifosine应用于胶质细胞瘤的Ⅱ期临床试验仍在进行,预期于2020年5月结束。
2
变构抑制剂变构抑制剂
不直接作用于ATP结合位点或PH结构域,而是结合在PH结构域与激酶域连接处一个独特的疏水区域,稳定Akt“PH-in”构象,封闭磷脂结合位点,阻断Akt的激活[21]。
Akt变构抑制剂最早由默沙东开发,通过高通量筛选找到5,6-二苯基-吡嗪-2(1H)-酮和2,3-二苯基喹喔啉两个结构系列,进一步衍生得到Akti-1、Akti-2及双重抑制的Akti-1/2三类变构抑制剂[22]。
但Akti-1/2的溶解度和药动学性质较差[23],随后在其基础上研发了MK-2206,对Akt1、2和3体外抑制的半数抑制浓度(IC50)分别为8、12和65nmol·L-1[24]。
此外,进入临床试验阶段的变构抑制剂还有miransertib(ARQ-092)、ARQ-751、BAY-1125976和TAS-117等。
MK-2206是临床研究最多的Akt变构抑制剂,但目前已不再列于默沙东的研发管线中。
早期对乳腺癌患者的一项术前研究中,MK-2206就被报道出较多的Ⅲ级不良事件。
尽管给药剂量不同,但入组的12例患者中有一半出现了皮疹或皮肤瘙痒[25]。
一项Ⅰ期临床试验确定了MK-2206的最大耐受剂量(MTD)是隔日60mg,剂量限制性毒性(DLT)为皮疹和口腔炎,不良反应有皮疹(51.5%)、恶心(36.4%)、皮肤瘙痒(24.2%)、高血糖(21.2%)和腹泻(21.2%)[26]。
一项MK-2206单药治疗淋巴瘤的Ⅱ期临床试验入组59例患者,初始剂量为200mg,每周1次,根据患者的耐受程度调整用药剂量,28d为一个治疗周期,未出现疾病进展或严重不良反应最多给药12个周期。
33例患者最终剂量达到每周300mg,8例患者减少至每周135mg,客观缓解率(ORR)为14%,有53%的患者出现皮疹,其中15%为Ⅲ级不良反应,患者血清细胞因子检测结果显示部分细胞因子(如白细胞介素10等)增加,可能为抑制靶点引起的负反馈调节所致[27]。
另一项评估MK-2206治疗胃癌和胃食管结合部癌的Ⅱ期单臂临床试验中给药剂量为隔日60mg,结果显示70例患者缓解率仅1%,中位PFS为1.8个月,中位OS为5.1个月,43%的患者出现了皮疹的不良反应[28]。
在与现有肾细胞癌靶向药物依维莫司的Ⅱ期双臂对比试验中,43例抗血管内皮生长因子(VEGF)治疗耐药的晚期肾细胞癌患者按2:1随机分为MK-2206组(n=29)和依维莫司组(n=14),MK-2206组给予MK-2206200mg,po,每周1次;依维莫司组给予依维莫司10mg,po,qd。
MK-2206组和依维莫司组中位PFS分别为3.7个月和6.0个月,MK-2206疗效并未优于依维莫司,反而增加了诱发皮疹的概率[29]。
MK-2206与厄洛替尼联用治疗厄洛替尼耐药的非小细胞肺癌,厄洛替尼每日150mg,MK-2206隔日45mg,连续给药12周。
研究结果表明,80例患者中45例EGFR突变患者的总缓解率为9%,35例EGFR野生型患者的总缓解率为3%,低于20%的预期目标[30]。
而另一项MK-2206联合MEK抑制剂selumetinib治疗结直肠癌的Ⅱ期临床试验中,21例入组患者未出现客观缓解[31]。
MK-2206也不能增强阿那曲唑(anastrozole)用于PIK3CA突变、雌激素受体(ER)阳性、人类表皮生长因子受体2(HER2)阴性乳腺癌患者的治疗效果[32]。
3
ATP竞争性抑制剂
该类抑制剂结合激酶域ATP位点,表现为ATP竞争性抑制。
但因AGC激酶家族ATP结合位点结构高度相似,寻找专一的激酶抑制剂并不容易。
同时,Akt3种亚型激酶域的序列同源性达80%以上[13],当前进入临床阶段的ATP竞争性抑制剂均无法区分Akt各亚型,研发选择性抑制Akt特定亚型的ATP竞争性抑制剂也是未来致力于实现的目标。
已进入临床阶段的部分ATP竞争性抑制剂概况及主要缺点见表1所示。

3.1 早期ATP竞争性抑制剂
GSK-690693是第一个进入临床试验阶段的ATP竞争性抑制剂,对Akt1、2和3体外抑制的IC50分别为2、13和9nmol·L-1,也抑制PKCη、PKCθ和PrkX(IC50分别为2、2和5nmol·L-1)等[33]。
但其口服生物利用度低,Ⅰ期临床试验未完成就因严重不良反应(特别是高血糖)停止了研发[34]。
AT-13148对Akt1、2和3体外抑制的IC50分别为38、402和50nmol·L-1,同时也抑制p70S6K、PKA和ROCK1/2(IC50分别为8、3和6/4nmol·L-1)等[35]。
AT-13148临床试验开始于2012年,部分公开数据显示,入组晚期实体瘤患者30例,每周一、三、五给药3次,剂量5~240mg,MTD为240mg,常见的不良反应有低血压、恶心、厌食和头疼[36]。
目前未见相关研究数据更新。
LY-2780301由礼来开发,是Akt和p70S6K的双重抑制剂。
Ⅰ期临床试验确认了MTD为每日500mg,不良反应有便秘(19%)、疲乏(13%)、恶心(9%)和腹泻(9%)[37]。
一项LY-2780301联合吉西他滨用于PI3K/Akt/mTOR信号通路异常的晚期或转移性实体瘤患者的临床Ⅰb期剂量爬坡研究表明,其不良反应可控,5%患者出现部分缓解(PR)[38]。
此外,一项LY-2780301联合紫杉醇治疗HER2阴性乳腺癌的Ⅰb/Ⅱ期临床试验尚无后续报道,LY-2780301当前状态为临床Ⅱ期研发停止。
M-2698(MSC-2363318A)由默克雪兰诺公司开发,也是Akt和p70S6K的双重抑制剂。
Ⅰ期临床数据显示,剂量爬坡(15~380mg·d-1)与剂量扩大(240mg·d-1,连续21d)研究均显示患者耐受性良好,未达到MTD。
12周后,32%的晚期肿瘤患者临床获益,但没有观察到肿瘤完全缓解(CR)或PR[39]。
目前M-2698的研究状态仍处于Ⅰ期临床研究中。
3.2 在研ATP竞争性抑制剂
3.2.1 ipatasertib(GDC-0068)
ipatasertib是现阶段临床研究进展最快的ATP竞争性抑制剂,由ArrayBioPharma开发,现由罗氏与中外制药合作推进。
对Akt1、2和3体外抑制的IC50分别为5、18和8nmol·L-1[40]。
ipatasertib的优势在于其独特的、具有手性的嘧啶并环戊醇结构,使其对Akt的抑制是对同源蛋白PKA抑制的620倍,且对另外230种体内常见激酶几乎无抑制。
ipatasertib对前列腺癌、乳腺癌和卵巢癌等多种移植瘤模型均显示出良好的抑制效果[41]。
ipatasertib的一项Ⅰ期临床试验确定了MTD为600mg,给药周期为28d,前21d连续给药,qd,后7d停药。
安全性方面,92%的患者用药后出现了至少1种不良反应,600mg剂量以下无Ⅲ级以上不良事件,常见的Ⅱ级以上不良事件有腹泻(35%)、恶心(27%)和乏力(25%),可通过支持疗法和控制剂量改善,患者可继续服用ipatasertib,证实其耐受性良好[42]。
ipatasertib联用紫杉醇治疗TNBC的Ⅱ期临床试验首次确认了ipatasertib对TNBC的疗效[3]。
124例TNBC患者随机分配至治疗组(ipatasertib400mg+紫杉醇)和安慰剂组(紫杉醇+安慰剂),每组62例。
研究表明治疗组中位PFS为6.2个月,比安慰剂组的4.9个月相比显著延长(P=0.037)。
对患者的肿瘤基因筛查显示,在PTEN表达较低的肿瘤患者中,治疗组的中位PFS为6.2个月,安慰剂组为3.7个月(P=0.18);PIK3CA/AKT1/PTEN基因突变的患者中,治疗组的中位PFS为9.0个月,安慰剂组为4.9个月(P=0.041);未发生PIK3CA/AKT1/PTEN基因突变的患者中,治疗组的中位PFS为5.3个月,安慰剂组为3.7个月(P=0.36)。
常见Ⅲ级以上不良事件有腹泻(23%)、中性粒细胞数偏低(8%)以及中性粒细胞减少(10%)。ipatasertib联用阿比特龙和泼尼松治疗转移性去势抵抗性前列腺癌(mCRPC)的Ⅱ期临床研究显示,对于PTEN基因缺失的患者放射影像学中位PFS由4.6个月提高到11.5个月(P=0.0064),而非PTEN缺失的患者也有部分提高,由5.6个月提高至7.5个月(P=0.5647)[43]。
但ipatasertib与mFOLFOX6化疗方案(奥沙利铂+亚叶酸钙+氟尿嘧啶)联合用于治疗胃癌和胃食管结合部癌的Ⅱ期临床试验研究结果显示,治疗组相较于对照组未实现临床获益。2017—2018年,先后开始了2项ipatasertibⅢ期临床试验,分别为ipatasertib联用阿比特龙治疗mCRPC以及ipatasertib联用紫杉醇治疗PIK3CA/AKT1/PTEN基因突变的TNBC与孕激素受体(HR)阳性、HER2阴性的乳腺癌,目前仍在招募患者中。
3.2.2 capivasertib(AZD-5363)
capivasertib由阿斯利康研发,对Akt1、2和3体外抑制的IC50分别为3、8和8nmol·L-1,对p70S6K和PKA抑制的IC50为6和7nmol·L-1[44]。
一项针对PIK3CA突变的乳腺癌和妇科肿瘤的Ⅰ期临床试验中,每日给药2次,连续给药(7/7)和一周前2日(2/7)给药的MTD分别为320mg和640mg,DLT分别为皮疹及腹泻和高血糖,而一周前4日(4/7)给药的MTD为480mg,未出现DLT。
常见的不良反应有腹泻(78%)和恶心(49%),Ⅲ级以上不良事件主要是高血糖(20%)。
该研究推荐Ⅱ期临床试验的口服给药剂量为480mg,每日2次,并采用一周前4日给药、后3日停药的方式。
结果显示给药后患者肿瘤体积有所减小,但缓解率不足10%[45]。
此外,capivasertib联合多西他赛和泼尼松龙用于mCRPC的Ⅰ期临床试验,推荐Ⅱ期临床试验的剂量为320mg,每日2次,一周前4日口服给药[46]。
capivasertib联合紫杉醇用于ER阳性的乳腺癌Ⅱ期临床试验,入组110例患者,治疗组和安慰剂组的中位PFS分别为10.9个月和8.4个月,ORR分别为59%和57%。
治疗组常见不良反应有腹泻(76%)、脱发(52%)和恶心(39%),Ⅲ级以上不良事件发生率治疗组和安慰剂组分别为59%和31%[47]。
2019年6月,capivasertib联合紫杉醇一线治疗晚期和转移性TNBC的Ⅲ期临床研究,预期招募800例患者,目前正在患者招募中,针对其他恶性肿瘤的Ⅰ/Ⅱ期临床试验也在进行中。
3.2.3 afuresertib
(GSK2110183)和uprosertib(GSK2141795)afuresertib和uprosertib由葛兰素史克开发,诺华收购葛兰素史克肿瘤业务后,转至诺华旗下,2018年,国内来凯医药获得了这2款抑制剂的开发权。
afuresertib对Akt1、2和3体外抑制的IC50分别为0.08、2和2.6nmol·L-1,uprosertib对Akt1、2和3体外抑制的IC50分别为0.066、1.4和1.5nmol·L-1,2款抑制剂对PKA抑制的IC50为1.3和2.0nmol·L-1[48]。
一项afuresertib用于血液肿瘤的Ⅰ期临床试验中,入组73例患者,给药剂量为每日25~150mg,该试验确定了MTD为125mg·d-1。
常见不良反应有恶心(35.6%)、腹泻(32.9%)和消化不良(24.7%)[49]。
另一项afuresertib联合MEK抑制剂曲美替尼治疗实体瘤和多发性骨髓瘤的Ⅰ期临床试验,入组20例患者,临床结果表明MTD为afuresertib25mg·d-1、曲美替尼1.5mg·d-1或afuresertib50mg·d-1、曲美替尼1.0mg·d-1。常见不良反应有腹泻(60%)、痤疮性皮炎(55%)和斑丘疹(45%)。
有效性方面,入组患者中仅1例实体瘤(黑色素瘤)患者出现PR[50]。
afuresertib联合奥法木单抗治疗慢性淋巴细胞白血病的Ⅱ期临床试验中,入组30例患者,中位随访13.4个月,总缓解率50%,其中CR率为3.6%,中位PFS为8.5个月,OS为34.8个月[51]。
一项评估afuresertib联合卡铂与紫杉醇治疗卵巢癌的Ⅰ/Ⅱ期临床试验,入组29例患者,2例CR,14例PR,总缓解率37%,中位PFS为7.2个月,Ⅲ级以上不良事件有中性粒细胞减少(25%)和皮疹(7%)[52]。
一项uprosertib的Ⅰ期临床试验入组了77例实体瘤患者,确定了MTD为75mg·d-1,有2例PR,常见不良反应有腹泻(64%)、恶心(61%)、疲乏(49%)、呕吐(43%)和食欲减退(39%)[53]。
目前来凯医药计划以uprosertib联合曲美替尼开发肿瘤相关适应证。
展望近年来医药界在提高生物标记物和抑制剂对激酶的选择性方面取得了重大进展,使Akt抑制剂临床研究的成功率上升,也使高选择性的ATP竞争性抑制剂成为研究热点。
国内首个进入临床试验阶段的Akt抑制剂HZB0071,由哈尔滨珍宝制药与药明康德合作开发,于2018年9月获得临床批件[54]。
未来Akt竞争性抑制剂的研究方向在于提高激酶选择性、降低给药剂量、扩大治疗窗口,以期发现疗效更好、更安全的临床药物,以及进一步探索Akt各亚型单独应用或与其他药物联用的方案。
参考文献
详见 中国新药与临床杂志 2019年10月第38卷第10期