
文末有2019-2020海外505(b)(2)项目清单。
导语
近年来,在带量采购等政策的影响下,仿制药很难再持续维持医药市场的主要增长点的地位,高销售费用向高研发投入的转变将成必然。然而,整体研发能力还有待提升的客观形势下,开发创新药物只是少数企业的“特权”。
伴随着老年化,慢性患者的数量持续增加,改良型新药可降低药物副作用,增加药物顺应性,对慢性病患者具有强大的吸引力。因此改良型新药,这种相对创新新药来说,高临床成功率,高收益,长生命周期的产品持续被市场看好。预计我国2025年新型制剂市场规模将达2760亿元,占药品市场比例能达到10%。
目录
1. 带量采购下的转型新路
1.1背景介绍
1.2申报数据分析
1.3风险和挑战
2. 505(b)(2)的快速崛起
3. 505(b)(2)和505(b)(1)申报审批比较
4. 505(b)(2)的优势
4.1高临床成功率
4.2满足临床/市场需求
4.3投入成本低
4.4可申请快速通道
4.5专利壁垒高,生命周期长
4.6灵活选择RLD
5. 中国药企可借鉴的案例和运行模式
5.1成功的商业价值
5.2成功的市场独占权
5.3成功的商务合作
6. 2019-2020活跃的海外505(b)(2)项目清单
带量采购下的转型新路
背景介绍
2019年8月20日发布的2019年版医保目录,一次性调出了150个品种,这150个品种2018年的终端销售额超过400亿元,随着2020年1月1日新版目录执行,迎来了大规模的品种踢出,市场将面临猛烈的冲击。被踢出的药品中,有一半是临床价值不高,有更好替代的药品。
一方面面临着被踢的风险,另一方面医保竞标的竞争也非常激烈,随着大批量产品通过一致性评价,国内药企之间的价格战只会越演越烈,依靠仿制药坐享亿万市场的时代一去不复返。最近的集采现场,华东医药20亿的大品种阿卡波糖口服缓释剂型不幸落标,股票直接跌停。
仿制药的价格和利润都将大幅下跌,为创新药进医保扫清了障碍,也倒逼药企走出仿制的舒适期,向创新转型。回顾美国市场的历史经验,梯瓦通过仿创结合的方式实现产业升级,特别是改良型新药的全面布局,为国内药企提供了可借鉴的经过市场验证的成功战略。
此外,对于改良型新药的政策鼓励也推波助澜:2016年《化学药品注册分类改革工作方案》首次提出2类改良型新药概念,并强调 “具有明显临床优势”。既能够避免不必要的低水平重复,又能实现针对创新研发的进一步细分。政策指引下,针对改良型新药开展的研究逐渐增加。
2017年,《“十三五”中医药科技创新专项规划》政策中指出要重点发展缓控释给药系统,靶向给药系统,基于新型纳米技术的释药系统、新型头皮给药系统等制剂新技术。
申报数据分析
2类新药按新药途径申报包括4类:结构改良(2.1类)、剂型改良(2.2类)、新复方制剂(2.3类)和新适应症(2.4类)。总览国内改良型申报数据,其中2.4类受理号(新适应症)最多,高于50% ,但大部分都是进口药物。国内企业申报的以 2.2 类为主(注射用前列地尔脂质体、尼麦角林缓释片等),其次是2.1类(左旋氨氯地平、异甘草酸镁、左奥美拉唑等),最少的是2.3类 (6.5%)。而2.3类对应的505(b)(2)的Type4,占FDA申报的12%(见下文)。
2.2 类改良型新药主要指含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,比如绿叶制药的紫杉醇脂质体。因为不改变药物的药理活性,并且通常不需要做大临床验证,只需要通过 BE 试验证明其药动学一致即可。在成本方面要小得多,这也符合我国目前企业的特点。
2.1 类和2.3 类改良型新药申报较少,核心原因是技术门槛相对较高。比如2.4类,需要考虑到药物之间的不相容、片重的比例、载药量限制等多个因素。典型的例子是葛兰素史克用于治疗 HIV 的单片复方制剂绥美凯(Triumeq),包含Tivicay、Abacavir、Lamivudine三种药物分子的复方制剂。其次这两类新药大概率会做大临床,以证明其疗效,研发时间长、风险相对较大。
风险和挑战
· 改良型新药是基于原研药,因此开发商通常会依赖之前申报的安全数据。但安全性不是一成不变的,因此要特别关注原研药物获批后的不良事件资料。
· 和505(b)(2)不同,2类药物特别强调要有明显的临床优势,除确定科学可行性之外,需要深入的思考产品是否有明确的优势,是否解决了临床的一个关键问题,这个关键问题是否可转化成市场的需求。
· 按照新注册政策,不再硬性规定2类药品的临床试验,需企业自己去把控,这对企业临床研究能力提出更高要求。这就要求企业和CDE保持良好的沟通,通过对获批案例的分析以及本身产品的特质,设计最佳临床方案,最大程度降低临床试验投入,加快研发速度,并及时关注政策的改动。
· 虽然2类药品临床周期相对比较短,但市场是变动的,所以建议带着发展的眼光去评估产品的商业价值。一些现在的重磅产品,是否在未来几年内会受到新入竞争产品的威胁?老年化趋势会造成怎么样的市场板块变化?有哪些缝隙市场值得探索?哪些临床数据能助力销售,特别是在带量采购如果能提高中标机会等。
· 一个成功改良型新药,可撬动百亿级别的市场(见下文),但前提是产品的技术门槛够高,复制难度高。对研发能力还处在成长期的中国企业而言,505(b)(2)产品引进不失为现阶段风险低,回报率高的首选方式。
· 在引进海外505(b)(2)时,部分无临床试验获批的505(b)(2)产品,根据2类药品申报的规定,需考虑到国内临床的成本,以及相应带来的风险。
505(b)(2)的快速崛起
在深入剖析505(b)(2)的优势前,先回顾一下历史沿革。
1984年,美国国会通过了《药品价格竞争和专利期修正案》(Hatch-Waxman Amendment),将505(b)(2) 和505(b)(1)作为两种新药申报途径。505(b)(2)申报数据可援引FDA已有结论,因此该路径下的改良型新药兼具“创新-仿制”特点。欧盟药品管理局(EMA)此类途径的命名更简单明了:混合式药物 (Hybrid Medicines)。
至此,505(b)(1)、505(b)(2)和简略新药申请 (ANDA)构成药物申报的三个途径。
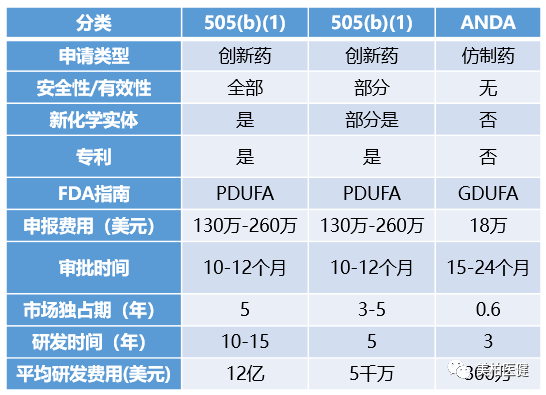
▲表一:三种申报途径的比较
和505(b)(1)不同,505(b)(2) 路径允许申请的数据部分或者全部来自于其他外部资源,比如公开发表的文献及FDA对上市药品安全、有效性等结论,通常来自于药品各项审评的结论。
修正案颁布以后,505(b)(2)的申请和获批并没有大幅度的提高。一直到90年代初,获批数量开始缓慢增长,2004年迎来了井喷,第一次超过了505(b)(1)的数量。

有意思的是1984年修正案颁布后,仿制药申报和获批有了大幅度增长,很大一个原因是1962年以后获批药物首次成为了合格的可仿制原研药,在仿制药依旧有市场竞争力的情况下,开发商更倾向这种熟知的途径。
随着时间的流逝,仿制药市场竞争日趋激烈,促使开放商必须要扩大野心,不仅仅满足于复制原研药产品。近期的数据显示,大型仿制药开发商的平均市值从2016年的80亿美元降低到了2019年的50亿美元。同时,随着技术的发展,特别是新型制剂的更新换代,如纳米技术、智能给药和新型功能材料等,为505(b)(2)的开发奠定了技术基石。
此外,2010年到2015年期间,由于专利保护的损失,原研药产品的收入损失了1200亿美元。另一方面,505(b)(1)监管的要求日趋严格,增加了临床试验的难度、需要更复杂的程序、更严格的入组患者标准、更详细的毒副作用报告,导致药企需投入更多的时间和金钱成本,回报率也从10%左右降低到3.2%。
再加上一系列的监管改革,比如获得非专利市场专有权,申请快速通道等也一齐助力505(b)(2)的蓬勃发展。
505(b)(2)和505(b)(1)申报审批比较
505(b)(2)申请类型一般包括以下几种:新剂量(Type 3);复方产品 (Type 4); 新配方或其他差异(Type 5);新适应症 (Type 6); 处方药变非处方药 (Type 8), 在少数情况下,也包括新化学实体。从2013-2018年的FDA申报数据来看,Type5为主要的方式,占46%。

落脚到领域,中枢神经系统、传染病、心血管、肿瘤等为热门领域。
505(b)(2)在审批程序上和505(b)(1)采用同一套基本流程,但在步骤和每个环节的着重点上有一定的区别:

▲505(b)(2)审批途径
图片来源:Carmago 官网
科学顾问委员会
505(b)(2)的申请者需建立一个独立的外部科学顾问委员会,以帮助开展援引文献的质量评估工作。
Pre-IND
505(b)(1) 通常是进行药理、药代动力学、毒理学试验等并制定临床方案后,再进行Pre-IND会议,以获取临床的同意。但505(b)(2)是先进行Pre-IND会议,一方面获得FDA对此途径可行性的认可,另一方面是通过咨询获取最佳开发方案,然后再进行药品配方的开发和非临床试验。
CMC
对于505(b)(2) 产品,用于临床一期研究的临床试验材料(通常是临床生物等效性)必须代表商业制造过程,需要准备三个稳定性批次。因此,在启动临床研究之前,必须投入大量的CMC工作。
申报要求
· 如果Cmax 和AUC与已批药物相同,需提供最低毒理数据
· 在大部分情况下(超过50%),只需要提供单剂量的BA / BE研究
· 当BA和已批药物不同时, 如缓释剂等,需额外临床来证实安全性/药效
· 如改变给药途径,需要提供对靶向器官的的毒性数据
· 新的适应症/针对特殊人群(儿科药)需进行临床试验以确保安全性

▲图片来源:Ther Innov Regul Sci. 2019 Jan7:2168479018811889
505(b)(2)的优势
高临床成功率
和505(b)(1)相比,研发时间成本大大的降低(见表一),除此之外,最新数据显示505(b)(1)的开发成功率只有13.8% (Wong et. al),肿瘤相关的药物成功率更低, 为3.4 %,而Carmago公开数据显示,505(b)(2)的成功率有22.5%。从上文数据来看,CNS和肿瘤这些505(b)(1)成功率相对较低的领域占505(b)(2)获批的重要比例。

▲ 表二:新药开发成功率
满足临床需求
早期药企为了充分利用化合物专利期,新药的开发往往很难同时解决溶解性、稳定性、吸收、代谢等多方面的问题,而505(b)(2)则在此基础上能达到:
· 提高药物效果:亮丙瑞林微球结构使药物活性成分不易被酶降解,改善药物稳定性,显著提高药物效果
· 减少用药次数,增强患者顺应性:1992年拜耳公司研发出高科技控释剂型硝苯地平控释片,开创了高血压药物治疗“1日1次”的全新模式。
· 降低副作用:紫杉醇白蛋白纳米粒采用纳米粒技术,将药物结合于人血白蛋白形成直径为130nm的颗粒,过敏反应发生率极低,血液毒性、消化道毒性及神经毒性均低于紫杉醇注射液。
投入成本低
(见表一)
可申请快速通道
和505(b)(1)类似,505(b)(2)也可以申请快速通道途径,进一步加速获批的时间,如下图所示,Priority为最常见途径。

专利壁垒高,生命周期长
和仿制药比较(首仿的市场独占为180天),一般来说,FDA会授予505(b)(2) 3年的市场独占,若申请项目是新化学实体,为5年,孤儿药为7年,儿科药的话则在保护期末尾再加6个月。

灵活选择RLD
根据Paragraph IV规定,ANDA途径有可能会面临30个月的诉讼,但505(b)(2)选择参比制剂(RLD)更有灵活度,可有效的规避,加速产品上市。比如Hikma因为选择了Col-Probenecid作为RLD,申请505(b)(2)途径,当其他仿制药开发商正在焦头烂额等待RLD拥有者武田的专利诉讼时,Hikma的的产品Mitigare在武田察觉前就争取到获批。
中国药企可借鉴的案例和运行模式
成功的商业价值
505(b)(2)产品和原研药相比,有着临床的优势,因此给开发商带来了巨大的商业利润。从销售峰值来看,同一时期的505(b)(2)产品平均峰值销售额在9000万美元至2.7亿美元之间,总体平均销售额在2亿美元左右,证实了 505(b)(2)对市场的吸引力。强劲的销售和低廉的开发成本可带来可观的投资回报率。

除了销售额,市场份额也是很重要的一个评判指标。下表例举的几个例子来看,成功505(b)(2)的产品可占整体市场份额的70%以上。

▲表三:505(b)(2)上市稳定后市场份额
分解到个案(长效胰岛素等生物类似物不在讨论范畴):
· RAVICTI®(甘油苯基丁酸酯)于2013年获得批准,可用于不能通过饮食限制和/或补充剂治疗的尿素循环障碍(UCD)患者的慢性治疗。与参考药物BUPHENYL®(苯基丁酸钠)相比,RAVICTI提供了更大的给药便利性,从上市到2018年,其销售额达到8.24亿美元。
· VASOSTRICT®(加压素注射液)于2014年获批准用于增加患有血管舒张性休克(例如,心脏切开术后或败血症)的成年人的血压。到2018年底, VASOSTRICT的总销售额达到12.93亿美元。
· BENDEKA®(苯达莫司汀HCI)与参考产品TREANDA®相比,提供剂型的便利性,还包括易于保存且提供多种剂量选择,于2015年12月被批准为孤儿药。并在2016年至2018年期间实现了19.59亿美元的销售额。BENDEKA带来的销售额外,给开发商也带来了巨额利益,在后文中会继续讨论。
· 阿斯利康的Vimovo(萘普生/埃索美拉唑)提供了一种新的药物与埃索美拉唑的缓释组合,从而降低了萘普生产生胃溃疡的风险。年销售额的最高峰值约为6.12亿美元。值得指出的是,因为价位比单买两种药高很多的原因,后期的销售有受影响。
· Suboxone 是通过生物等效开发的口腔薄膜片,解决舌下片用药后口腔不适的问题,同时携带方便。口腔薄膜上市后,便将舌下片撤出市场。市场上销售额达到16亿美金。
成功的市场独占权
· 新化学实体:非氘代丁苯那嗪tetrabenazine(以商品名Xenazine出售)在2008年获得FDA批准。Austedo,丁苯那嗪的氘代版本,2017年4月通过505(b)(2)途径获批。氘版药物具有明显的较低的新陈代谢率,因此有更长半衰期。但和参考药物比较,因为替换了两组氢氘,成功的被列为新化学实体(NCE),可享用5年市场独占权。

· 规避首仿的180天独占权:辉瑞的重磅炸弹高血压药物Norvasc(苯磺酸氨氯地平)1992获批。2003年,Dr.Reddy也想将自己的仿制药进入市场,但已被另外一家公司抢了首仿的先机,按照规定,180天内其他的仿制药都不能进入市场。Dr. Reddy便提交了Norvasc的一种改盐型制剂amlodipine maleate,走505(b)(2)途径,绕过了180天的市场专有权,争取了提早进入市场的优势。
· 两种非专利药的新组合:Aralez Pharmaceuticals的Yosprala是阿司匹林和抗血小板药奥美拉唑质子泵抑制剂的组合,2016年获批后申请到涵盖此组合的专利(9,539,214),橙皮书中列出直到2033年才到期。而2014年获批的默沙东的复方抗生素产品Zerbaxa除了五年的新化学实体专有权,因为是针对传染病,拥有额外的5年,共计10年市场专有权。
· 延长产品生命周期:奥美拉唑Prilosec在美国1989年上市时,获得了超过50亿美元的年销售业绩,为了延长药物生命周期,阿斯利康开发了埃索美拉唑Nexium,2001年获批后2005年便达到50亿美元的峰值年销售。Nexium成功的关键之一在于开展了大量的临床试验,而不是单纯的生物等效实验,并通过在临床实验中与Prilosec巧妙的剂量对比找到了卖点,从而一跃成为重磅产品。另一个典型的案例就是Abbott的降胆固醇的药物非诺贝特,最开始是Tricor-1,快批下来时,开发Tricor-2,紧接着又是Tricor-3。因为非诺贝特是体内分解产物在起作用,把分解产物做成药物开发Trilipix,继续占据市场。

▲图片来源:Arch Intern Med. Author manuscript; available in PMC 2013 Apr 26.
成功的商务合作
基于505(b)(2)产品的合作分为两种,第一种是技术平台的“共生”合作,第二种是单一产品的合作。
上世纪90年代,伴随着药物释放技术的发展,全球诞生了350多家企业专门从事新型制剂的开发,较知名的有Alza公司(凭借Osmotic Pump® 这一个技术获得FDA的15个NDA,最后以105亿美元被强生收购)、Skyepharma(拥有Geomatrix®,Geoclock®和Soctec®三个口服控释平台,以适应患者的给药时间表或调整API摄入量,2016年Vectura以6.2亿美元收购SkyePharma)。
第三个案例不归属505(b)(2),但值得一提。Halozyme的核心技术Enhanze能够降解一类叫做hyaluronan的多糖,这类多糖暂时可逆降解后可以增加生物大分子的扩散吸收,进而让很多需要静脉滴注的药物改成肌肉注射。利用这个技术平台,和罗氏,辉瑞,BMS等多个国际药企达成了战略合作。
这些企业采用“共生式”商业模式,利用自己专有的技术平台,与专利药企业共同开发新型制剂新药,进行技术转让。
再举几个单一产品的合作案例:
· 2011年5月,Teva以68亿美元收购 Treanda,销售额一度在2014年增长到7.67亿美元。然而,Teva独占市场的好日子并不长久,2015年Eagle制药以同样的药物活性成分研制出了Bendeka并向FDA申报505(b)(2)的NDA。情急之下,为了保住市场垄断地位,Teva索性买下了Bendeka在美国的独家销售权,加上3000万美元的首付,该项交易金额最高可达1.2亿美元。Treanda在之后的几年里,逐渐失去对市场的掌控,被Bendeka抢占。
· 2018年10月,德国制药公司Grünenthal宣布以9.22亿美元的价格,接手阿斯利康拥有的Nexium(埃索美拉唑)欧洲市场的商业化权益,及对Vimovo除美国和日本地区外的全球市场的商业化权益。上文中已经提到过,在这次合作前,Vimovo已经给阿斯利康带了巨额利润。
· Tecfidera(dimethyl fumarate,富马酸二甲酯)是渤健的一款重磅多发性硬化症MS药物,2017年,Tecfidera销售额为42.14亿美元,为了稳固MS市场的龙头地位,同一年渤健和Alkermes就升级版口服富马酸药物Vumerity达成合作,和Tecfidera相比,Vumerity能改善的胃肠道耐受性。Alkermes将收到2,800万美元的预付款,以及最高总价值为2亿美元的里程碑付款。2019年,Vumerity通过505(b)(2)途径获批,包含了将Vumerity和Tecfidera进行比较以建立生物等效性的药物动力学桥接研究数据,并部分基于FDA对Tecfidera的安全和疗效结果。
2019-2020活跃的海外505(b)(2)项目清单
