
在过去的十年中, RNA-seq已经成为全转录组范围内分析差异基因表达和mRNAs差异剪接的重要工具,被应用于研究RNA生物学的方方面面,包括基因表达、翻译(翻译组,translatome)、RNA结构(结构组,structurome)、调节性RNA、RNA表观遗传学以及RNA动力学等。随着下一代测序技术的革新,RNA-seq技术也得到了不断发展,其应用领域也得到了不断拓展,例如空间转录学(spatialomics)等。加上近年来长读长测序和直接RNA-seq(direct RNA-seq)技术的应用以及数据分析计算工具的进一步整合,RNA-seq技术的创新使人们对RNA生物学有了更全面的理解。
回顾RNA-seq技术的十余年,自其诞生之日起,RNA-seq就成了研究分子生物学的普遍工具,在一定程度上奠定了我们对基因组功能的认知基础。但总地来说,RNA-seq中最常用的分析方法就是找出差异基因表达(Differential gene expression, DGE)。从始至今,虽然RNA-seq经常被应用于各类不同的研究中,但是DGE分析仍然是RNA-seq最主要的应用场景,并被视为常规研究工具。
科学研究的突破通常都是基于技术手段的突破更新,事实上,在RNA-seq之前,基因芯片技术曾是基因组表达分析的中坚力量。在这一技术“最辉煌”的时期,一提到基因表达研究,人们就会想到使用芯片。芯片技术简单、方便、快捷,能较准确地找到研究者需要找到的基因组信息。这些优点使它曾经风靡一时。然而与RNA-seq相比,芯片技术的局限性就显而易见了:
1. 芯片技术须依赖已知的基因组信息,在探索性研究和非模式生物的研究中存在局限性;而RNA-seq在这方面优势明显,它不需要预先设计探针,因此得到的数据集是无偏倚的,实现了无假设的实验设计[1,2],为未知转录本和变异发现研究提供了强有力的工具,这是芯片无法实现的。2. 芯片技术动态范围较窄和灵敏度低,芯片技术的动态范围通常为3个数量级(10³),而RNA-seq的动态范围要比芯片技术高几个数量级,可跨越5个数量级(>105) [1,3],因此,RNA-seq可检测的差异表达基因比例要高于芯片,特别是低丰度的基因 [3,4]。 3. 芯片技术通常无法完成对选择性剪接位点和新型异构体以及非编码RNA的检测,而RNA-Seq除了基因表达谱分析,还能鉴定选择性剪接异构体、剪接位点和等位基因特异的表达,且所有这些都在单个实验中完成。 因此,RNA-seq的出现很快就成为了RNA研究的主流手段。
时至今日,RNA-seq已从标准方法中延伸出多达约100种的RNA-seq方法,在RNA文库构建上也有了改良,如发表在Nature Methods上的以mRNA 3’端序列构建文库的方法,其采用的是Lexogen公司的QuantSeq文库构建试剂盒[5]。除此之外,以Illumina为代表的短读长测序平台能对大部分由不同方法构建的文库进行测序。另外,近几年长读长RNA-seq以及直接RNA-seq的进步也弥补了短读长测序无法解决的问题,帮助研究人员在如基因融合、结构变异以及不同剪切体上有了更加深入的了解。
那么今天,就让我们从多个维度共同了解一下常规RNA-seq与3’mRNAseq之间各自的优劣势,以便在今后的研究中选择更有利的方法进行研究。
一、流程
常规RNA-seq我们以典型的Illumina TruSeq RNA建库试剂盒为例与QuantSeq 3’ mRNA建库试剂盒进行比较。在常规的RNA-seq测序流程中,通常会包含以下几个步骤, mRNA富集或去除核糖体RNA(这里需要跟RNA的质量以及研究方向进行相应选择,对于降解的RNA样品,如RIN<7的样品应选择去除核糖体RNA),RNA片段化,第一链cDNA合成,第二链cDNA合成,加A尾加接头,文库扩增这些步骤,当然中间会有纯化的步骤(图1 左)。在常规RNA-seq中,通常需要1ng-2μg的total RNA input量。然而QuantSeq通常比标准RNA-seq法流程更为简单且需更低的total RNA input量:100pg-1μg。在QuantSeq流程中,每个转录本只产生一个片段,因此数据量是常规RNA-seq的1/10,并可以配合UMI模块对第二链cDNA进行单分子标签标记,使基因表达定量更加精确。QuantSeq采用oligo dT引物特异逆转录含poly(A)尾mRNA,第二链使用随机引物进行合成,随机引物结合的位置与poly(A)之间的距离决定了插入片段的长度,因此不需要poly(A)富集及片段化这一步骤,并在cDNA合成后立即进行PCR,从而取代了接头连接步骤使整个建库流程时间大大缩短。这种方法可以在低测序深度上实现与标准RNA-seq同等的灵敏度水平。由于QuantSeq中每个样本需要的数据量小,因此,这种方法可以实现更多个文库的混合同步测序(图2中和右)。
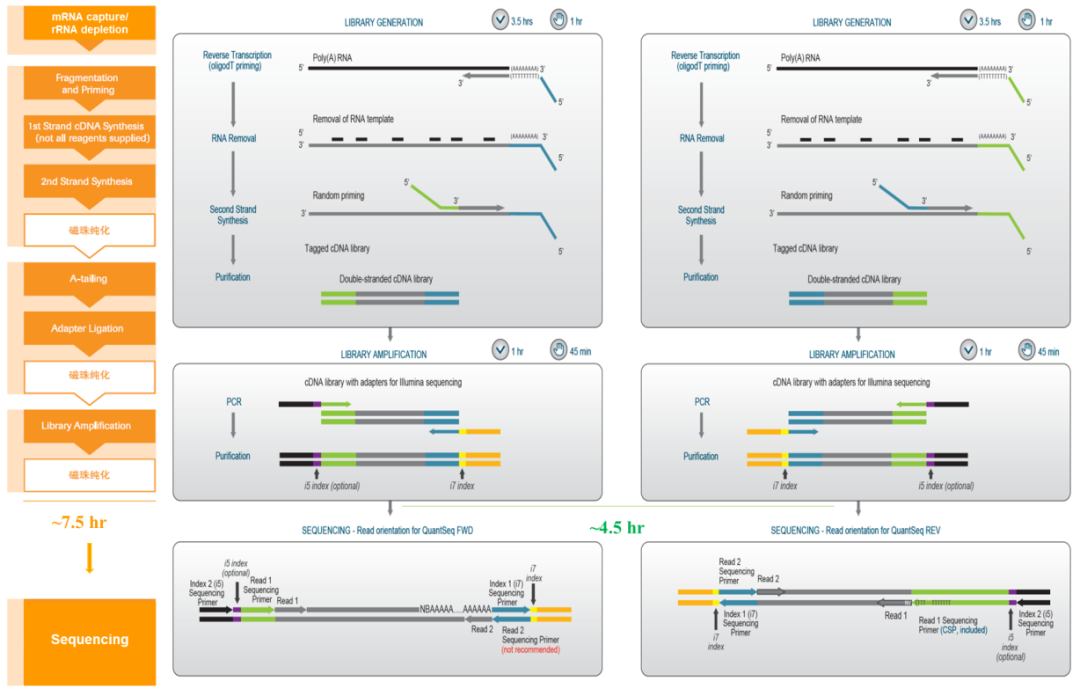
图1. 左:为常规RNA-seq建库测序流程,中间和右边分别为QuantSeq 3’ mRNA FWD和REV建库测序流程。中:QuantSeq FWD试剂盒Read 1(从绿色P5接头部分开始)测序对应的是靠近mRNA 3’端序列,可以使用Illumina测序引物进行测序,且费用较低。右:QuantSeq REV 试剂盒测序位置是 Read 1 和Read 2 的互换,Read 1 能够直接检测到转录本的末端。QuantSeq REV Read 1测序需要定制化的测序引物(CSP,包含在试剂盒内)
二、应用方向
在常规RNA-seq应用中最主要的当然还是以DGE分析为主,通常每个样本会测20-30 M的reads数进行高质量的DGE分析。此外,由于常规RNA-seq对整个转录本的序列进行打断后测序,其覆盖了转录本的完整信息(图4),因此除了最主要的DGE分析外,它可以进行转录本的de novo组装,Isoform的检测、定量以及基因融合的分析(图2)。对于后几项的应用,它们对数据量上有很大的提升要求,如Isoform检测需70-80M的reads每个样本[6],全转录组则需100M的reads数每个样本[7](图3)。相对于QuantSeq 3’mRNA seq,其富集的是mRNA3’ CDs以及UTR区域(图4),每个转录本只产生一个片段(图2),因此,仅需很少的数据量就可以进行准确的DGE分析,通常为3-10M的reads数[8],仅为常规RNA-seq的1/10(图2和3),因此大大节省测序空间,允许更多样品的混合测序,大大节省了成本。此外,mRNAs的APA化会产生3ʹ UTR长度不等的异构体。对于一个特定的基因来说,它不仅产生了这个基因的多个亚型,而且由于3ʹUTR中存在着顺式调控元件,这也会影响该转录本的调控。因此,QuantSeq对APA的研究者们来说可用于更详细地研究miRNA的调控作用,mRNA的稳定和定位,以及mRNA的翻译。

图2. 常规RNA-seq与QuantSeq 3’ mRNAseq在应用上的区别
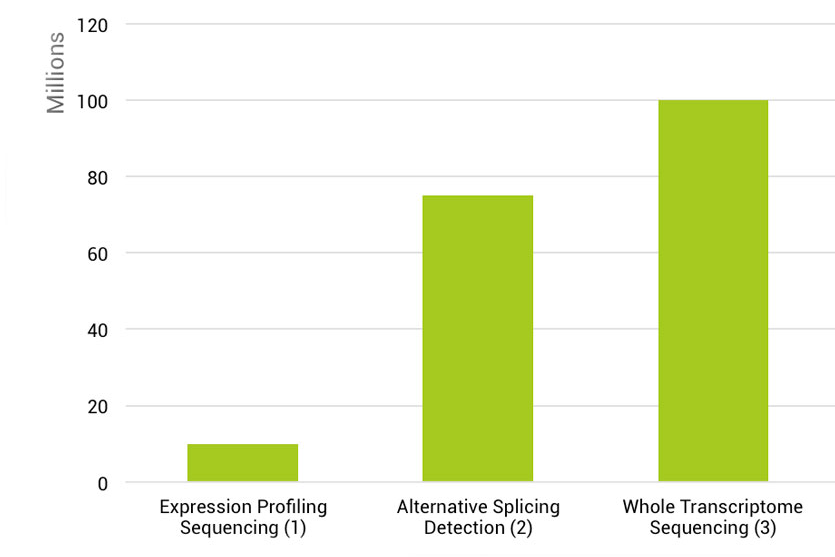
图3. 不同应用对数据量上的要求
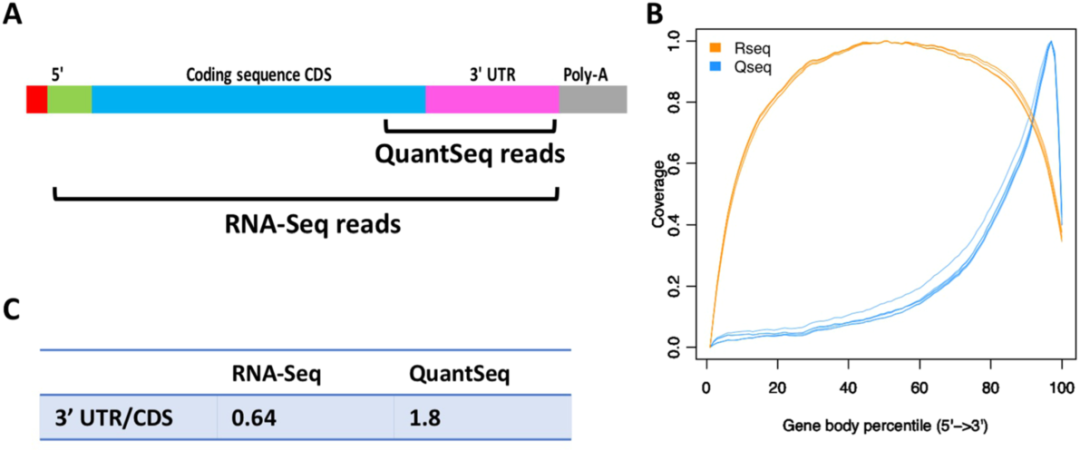
图4. 常规RNA-seq与QuantSeq 3’ mRNAseq reads在转录本的覆盖情况
对于isoform检测、定量研究,短读长的建库方案及测序平台存在着较大的局限性[9]。Weirather JL等人[9]通过采用金标准的Spike-in RNA标准品SIRVs E0(Lexogen)对PacBio、ONT以及Illumina测序平台对基因isoform检测性能进行了评估。E0模块包含来自7个人类模型基因的69种isoform等摩尔比例组成,综合反映了可变剪接、可变转录起始和终止位点、重叠基因和反义转录的变化(图5)。Weirather JL等人采用Lexogen提供的3种注释文库对不同测序平台进行了isoform检测性能评估,分别为“correct library”,注释包含所有68种真实表达的isoforms; “insufficient library”,注释仅包含68种真正表达isoforms中的43种;以及“over-annotated library”,注释含68个真实表达的isoforms和额外32个未表达的isoforms。3个注释文库,Illumina数据经StringTie进行isoforms重构,分别检测到44,63,62,其中分别有33,27,24假阳性预测;ONT测序直接检测到correct library文库中所有68个表达isoforms(表1),PacBio测序检测到67个,其中一个219bp isoform SIRV618因片段筛选时已过滤掉了。因此,相对Illumina,PacBio和ONT在isoforms检测中表现出超高优势。同时表明短读长拼接重构isoforms存在较大的缺陷,因此,常规RNA-seq较QuantSeq在isoforms检测中的优势也就显得没什么意义了。
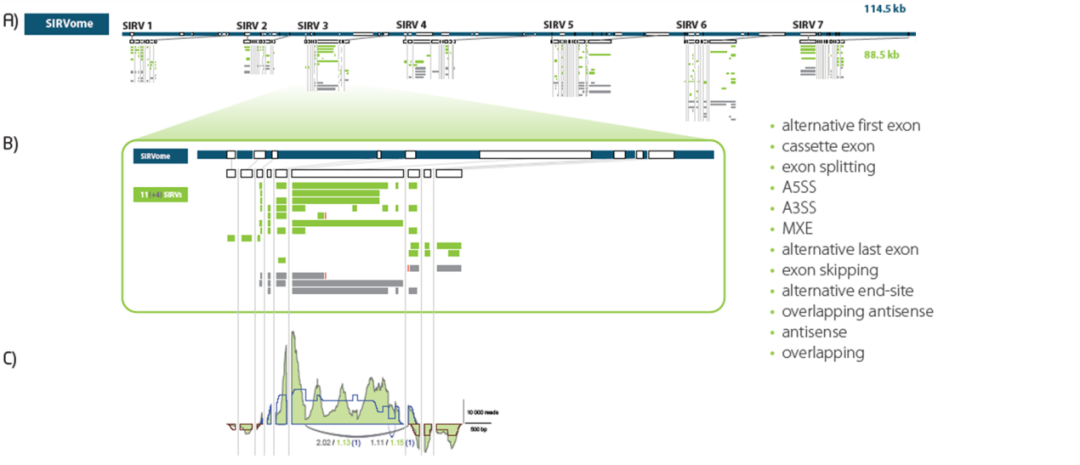
图5. SIRV设计概览。SIRV1到SIRV7,模拟人类模型基因,全面代表了主要的可变剪切方式以及重复和差异转录。A)7个SIRV基因的人工染色体,即SIRVome;B)SIRV3的放大图,提供11个转录本可变剪切体(绿色);灰色区域的转录本可变剪切体是附加的注释,用于其他的评估程序;C)SIRV mix中已知的转录本isoform浓度可以与预期的基因和外显子连接覆盖范围(蓝线为正链,红线为负链)与实验获得的reads覆盖范围(绿色区域)进行比较
表1. Illumina、PacBio和ONT在金标准SIRVs中isoform鉴定的表现
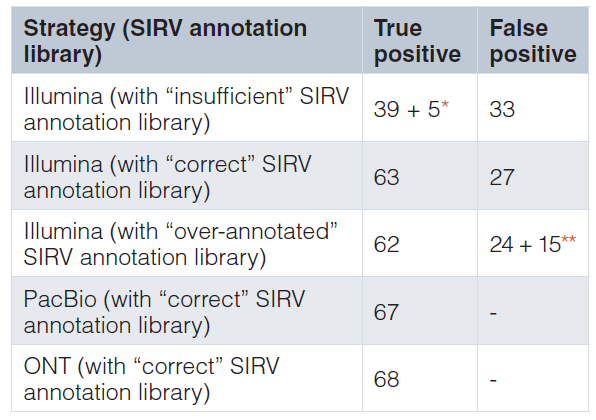
*在“insufficient ”的SIRV注释文库,其中有25种isoforms未被注释但表达。在这25个isoforms中,有5个isoforms在Illumina测序平台被检测到。
**在“over-annotated”的SIRV注释文库中,包含额外注释的32种isoforms,但没有真正表达。在这32种未表达的isoforms中,共检测到其中15个isoforms。
三、定量精确度
Moll, P.等人用ERCC spike-in RNA标准品的转录本覆盖reads数与input的分子数作图进行分析 (图6)。在线性模型评估和Spearman关联性评估中,QuantSeq展现出了非常高的input-output关联性和基因表达测定的准确性[9]。同时, 通过使用“erccdash- board” 软件,对QuantSeq和常规mRNA-Seq的差异基因表达检测能力进行了对比。当测定的读数(Reads)由10M降低到0.625M,QuantSeq维持了相当高的曲线面积值(AUC 0.860-0.897),而mRNA-Seq的曲线面积值较低,在0.736到0.776之间(图7)[9]。
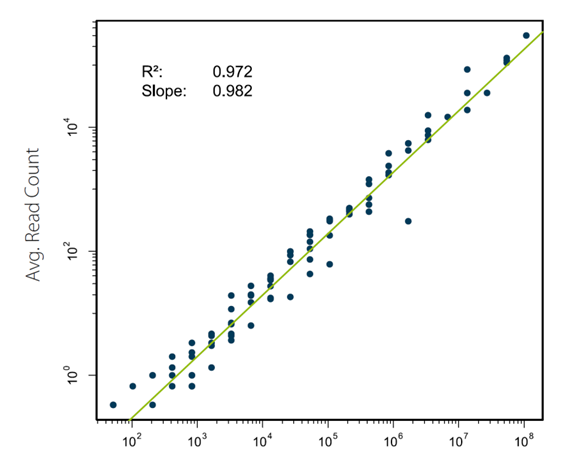
图6. QuantSeq-源于ERCC reads数与给定的input间存在极好的关联性
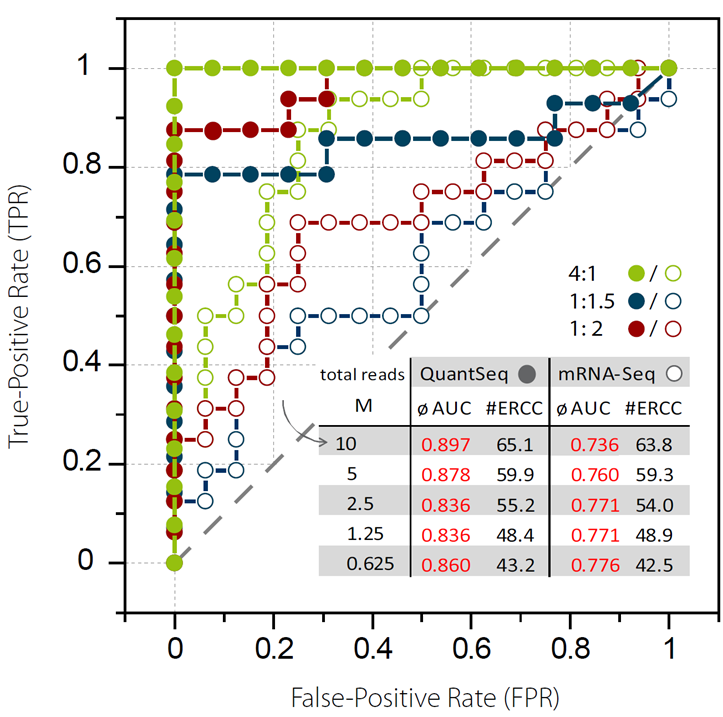
图7. QuantSeq和常规mRNA-Seq基因差异表达分析。给定的ERCC ExFold Spike-In Mix1和Mix 2间的倍数变化(4:1,1:1.5,1:2)用于评估真假阳性率(TPRs 和FPRs)。最优的基因差异表达检测体现在最大值为1的曲线面积(AUC)。用AUC对ERCC 检测到的RNA reads进行评估(测序数据量从10 M降到0.625 M)
四、低质量RNA中的表现
对于低质量,降解的RNA样本,如FFPE样本,常规的mRNA建库会导致3’端的偏好性,因此通常需要rRNA去除之后在进行建库,这样大大提高了建库的成本。而QuantSeq 3’mRMA文库构建本身就是以mRNA的3’端序列进行文库构建,主要集中在转录本的3’端,每个转录本只产生1个片段。这样能够使得无论RNA的质量如何(包含FFPE样本)都可准确定量。因此,相对于其他用Poly(A)分选mRNA操作流程,QuantSeq 3’mRNA-Seq更有效的对低质量的样本进行建库。
通过使用同一来源的不同质量RNA样本进行比较,评估QuantSeq适用于高度降解的样本(如FFPE样本)的能力。将人的MOLP-8肿瘤细胞系分成两份,一份进行新鲜冷冻,一份处理成FFPE样本,从而使同一个来源的样本得到不同质量的RNA。用RIN值(RNA完整度)来区分RNA的质量。RIN值大于8表示RNA质量高。对应严重降解的样本,RIN值不适用于质量的评估,因此使用DV200值(大于200nt的RNA片段的分布值)来表示RNA质量。低完整度的RNA对应低DV200值。RNA提取后,FFPE样本的DV200值为87%(RIN值2.8),冷冻样本RIN值为8.3。 使用50ng总RNA,用QuantSeq FWD 试剂盒进行文库构建。FFPE样本提取的RNA,即使DV200值低至23%(数据未显示)仍能成功构建QuantSeq文库。文库在Hiseq2500上进行用 1x 50 bp读长测序。结果显示FFPE-RNA文库和冷冻保存RNA文库的基因表达的相关性很高(R²= 0.86),表示QuantSeq能在不同质量的RNA中表现稳定(图8)。
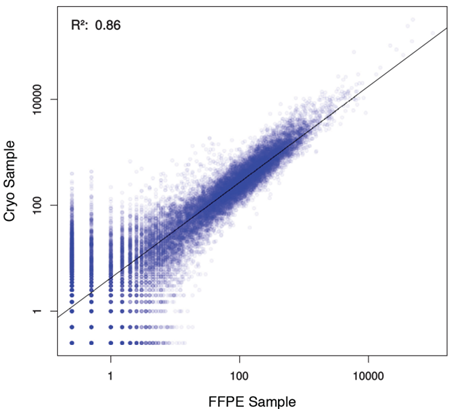
图8. FFPE和冷冻样本的基因表达的相关性
五、数据分析
常规的RNA-seq是以打断后的转录本进行建库测序,其覆盖的是整个转录本,因此与QuantSeq每个转录本只生成一个片段相比,其在数据上要远远大QuantSeq的数据量;另外,常规RNA-seq的数据在分析时需要将打断的转录本进行拼接以及RPKM的计算等流程,而QuantSeq无需复杂的计算,仅需基因片段的计数即可获得基因表达数据。因此,前者需要耗费大量的计算资源以及计算时间,而QuantSeq的数据分析就显得非常简单而省时了(6个样品35min内即可完成分析[9],现在会更快)。这对于讲究时效性的基于基因表达变化的辅助诊断及治疗来说是非常有利的。
此外,Lexogen与Bluebee® Genomics Platform达成战略合作,为QuantSeq 3’ mRNA-Seq建库试剂盒 (FWD 和REV)的数据提供免费分析。Lexogen为每个试剂盒提供一个密码,使用者可以登录该平台分析数据。也可用Partek Flow (license required) 平台进行数据分析。使用者可以直接在Partek Flow导入原始测序数据,然后进行自动化分析。也可进行定制化分析。
技术在不断更迭,在诸多的技术手段中找到最适合自己的研究往往就是成功的开始。QuantSeq 3’ mRNA-Seq在诸多方面都展现出了自己独特的优越性,特别在基因表达分析、3’UTR以及APA位点的分析等其他方面优势明显。因此,在关注于以上几点的RNA测序研究应用中,QuantSeq 3’ mRNA-Seq可谓不二之选。
Lexogen是一家专注于为RNA研究提供创新性解决方案的生物公司,产品线覆盖全面,从样本制备、RNA提取、RNA文库构建到最后的数据分析,Lexogen可提供完整的解决方案。其独特的链特异性文库构建流程,无需RNA打断,低input量要求,流程简单快速,仅需4.5h即可完成文库的构建,可大大节约时间成本。北京仲黎商贸有限公司作为贝克曼库尔特(中国)的战略合作代理商,同时也是Lexogen公司在中国的独家代理商。 欲详细了解QuantSeq 3’ mRNA-Seq及更多应用场景,可咨询sales@bjzltrade.com。