

单细胞测序技术使我们能够低成本、快速构建器官或者组织的大规模单细胞表达图谱,了解器官或组织内细胞复杂的基因网络以及单细胞中的调控机制,进一步探究各细胞群体的协同方式。2020年8月,来自康奈尔医学院的Dan Landau博士(同时也在纽约基因组研究中心任职)等人在著名期刊Nature Reviews Genetics上发表了一篇题为“Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics”的综述文章,为我们揭示了单细胞组学技术如何阐释肿瘤进化的奥秘。今天,就让我们通过本篇综述,深入了解如何利用单细胞测序这一新技术从遗传性和非遗传性两个角度审视肿瘤进化。
译者丨韩斐然 沈励泽
摘 要
癌症自身进化过程本身就是一个恶性肿瘤细胞群体本身不断“生长”不断增加的一个过程,导致其基因组信息越来越多样化,最终引起癌症不断复发,并对各种药物产生耐药性。 除了遗传因素外,细胞间的变异还包括了细胞本身状态、表观遗传学特征、空间分布及与细胞生长的微环境互作等因素,这些都会加剧和推动癌症发生演化。 因此,癌症的研究未来方向就是需要聚焦在单个细胞水平(体细胞进化最小单位)才能整合多个遗传维度。 这篇综述中,作者讨论了一些新兴的单细胞测序技术,能够为癌症进化提供一些新的线索。 这些信息表明,癌症是体细胞在遗传性因素和非遗传性因素之间复杂相互作用所共同导致的结果。
01
前 言
癌症的进化史包括恶性转化,然后发展为更具侵袭性和耐药性的形式,最终在临床上导致一些毁灭性的影响。体细胞突变,包括单核苷酸变异(SNV)和结构变异,对于癌症的发生和发展至关重要。但是近些年来,许多研究已经在健康组织中发现了大量的体细胞突变(somatic mutations),例如皮肤和食管暴露在环境致癌物后,组织中细胞发生突变,这表明癌症通常是由癌前克隆(pre-malignant clonal outgrowths)转化成的。重要的是,克隆的体细胞突变没有进展,甚至没有退化,与癌症的复发驱动因素重叠。这些数据表明,仅遗传机制可能不足以驱动肿瘤向着恶性程度发生转化。
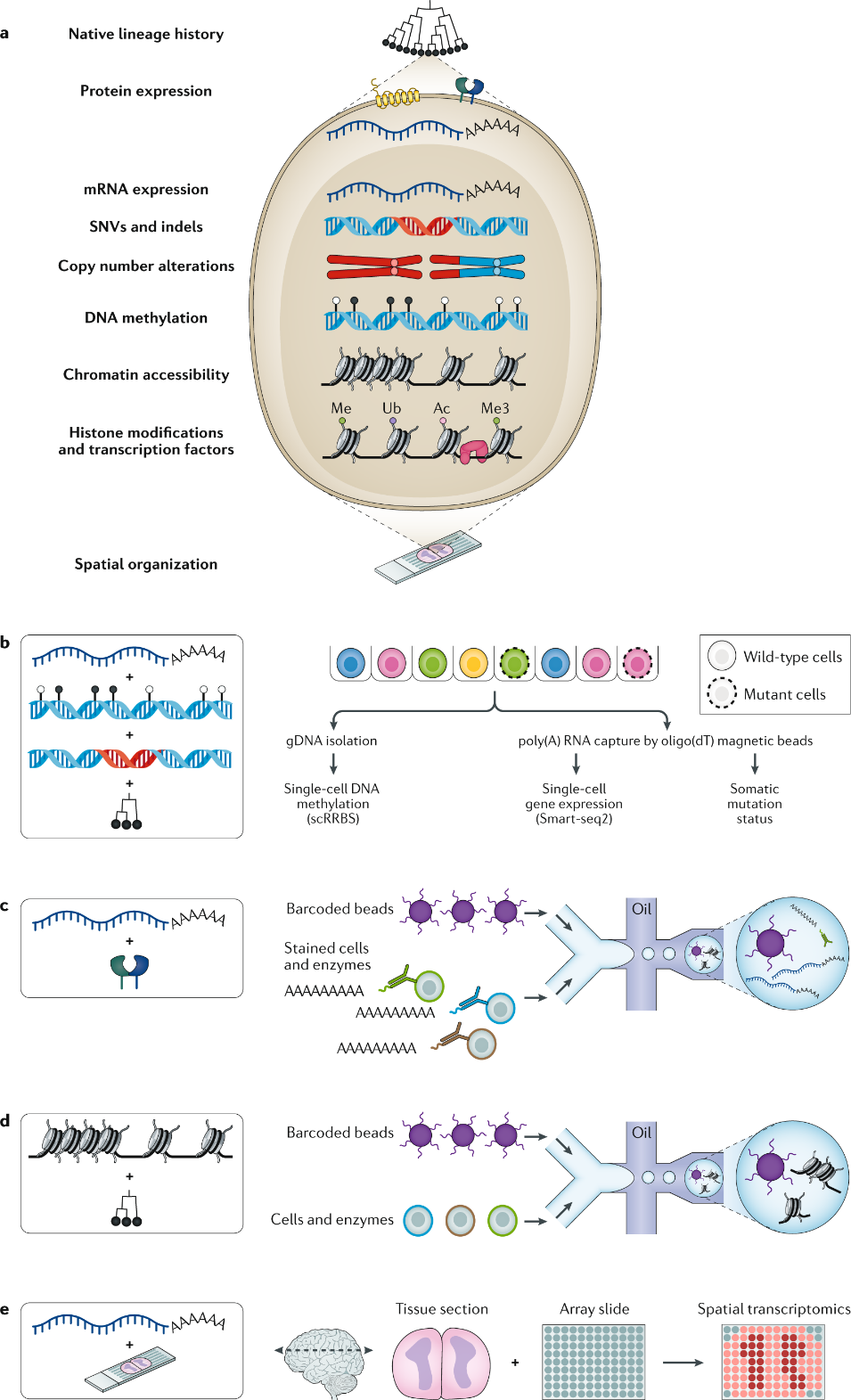
随着恶性肿瘤细胞种群数量的增长,细胞基因组在遗传水平会进一步多样化,从而使肿瘤发生进展和转移,并对药物产生耐药性。然而,大量研究证实并不是所有肿瘤进展、转移及耐药都与遗传驱动因素有关,所以这表明癌症进展有大量的非遗传因素参与。近几年兴起的多种单细胞测序技术如scRNA、scATAC、scMethy、scWGS、空间转录组等,分别揭示了细胞内肿瘤的异质性、表观遗传特征、组织空间动力学以及与肿瘤微环境的相互作用。这些技术可能能够为遗传性和非遗传性肿瘤内变异以及癌症的发展提供更多线索。因此,通过单细胞多组学技术能够整合单个癌细胞的多层信息,是全面理解癌症进化机制的关键。
本文中,作者回顾了单细胞技术在实验方面和生物信息方面最新的突破和进展,这些技术在单个肿瘤细胞中整合了可遗传信息的多个维度。这篇综述探讨了癌症进化的遗传和非遗传途径,这些途径可以通过单细胞多组学研究得到全新的认识。由于最近对癌症异质性的遗传来源和单细胞整合分析的技术方面的研究大量增加,因此作者在本文中呈现了大量令人信服的研究证据来表明,单细胞测序技术能够整合单个癌细胞中多种信息,从而破译肿瘤异质性与演化中的秘密。
02
遗传异质性与谱系追踪
Genetic heterogeneity and lineage tracing

2.1 Bulk测序对肿瘤克隆结构推断(Inference of clonal architecture in bulk sequencing)

通过持续积累(continuous acquisition)体细胞突变而产生的遗传异质性是许多癌症克隆进化的基础。这些遗传异质细胞群体的克隆结构先前已经从大量的二代测序结果中得到证实。
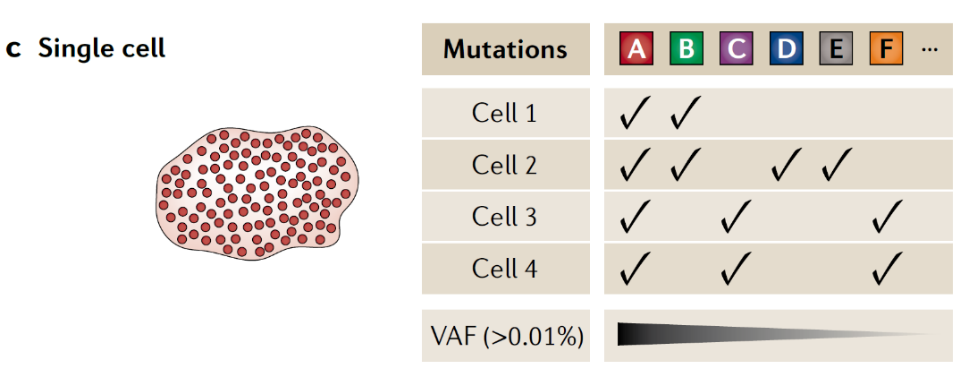
全外显子测序WES或全基因组重测序WGS数据中,整合体细胞突变的reads深度和变异等位基因频率可用于推断每个突变的肿瘤纯度、倍性和局部拷贝数,从而确定癌细胞分数(Cancer Cell Fractions, CCF)。这些数据可以在一定程度上解决克隆和亚克隆的关系。
在非整倍体实体瘤(aneuploid solid tumours)中,WGS可以通过确定SNV是否在大型扩增过程(例如全基因组复制WGD)之前或之后发生,来帮助对克隆体细胞的早期变化与晚期时间进行对比和时间上的推断。当分析大型队列时,发生驱动突变的进化序列的信息就能被揭示出来。
因此,驱动事件可以分为早期和晚期。早期经常是祖先突变或叫祖系突变(ancestral mutations),例如骨髓瘤中的DNMT3A和TET2突变等。晚期的亚克隆驱动,如恶性肿瘤细胞中的SF3B1,TP53和NPM1突变,会导致持续选择而产生的淋巴和髓样恶性肿瘤的突变。
· 知识拓展 ·
1. CCF
CCF即cancer cell fraction,简而言之,这个概念是指有多少细胞含有对应的变异。例如在某个肿瘤组织中所有肿瘤细胞都携带某个特定的体细胞突变,那么这个突变的CCF即为1。CCF可以通过VAF、肿瘤纯度和对应区间的拷贝数共同计算得到。
2. 非整倍体扩增肿瘤
基因组不稳定是肿瘤细胞的一个重要特征,非整倍体扩增指的是染色体数目并不是成倍增加,而是部分染色体随机丢失和获得,导致非整倍体的肿瘤细胞产生。这种现象普遍存在于各种恶性肿瘤细胞之中,对肿瘤的侵袭能力、耐药能力、转移能力等都有不同程度的影响。
3. 早期与晚期驱动事件
驱动事件按照在肿瘤发生发展过程中的时间顺序被(不严格地)分成了早期和晚期驱动事件。其中早期驱动事件通常可以在原发肿瘤中检出,一般对于肿瘤的恶变和发展起到驱动作用;晚期驱动事件则可能发生在部分亚克隆当中,影响肿瘤的表型改变、耐药和复发转移等过程。
同样,在肾细胞癌队列研究中,可以将肿瘤分为进化亚型。例如PBRM1中具有驱动事件的肿瘤,随后引起SETD2或PI3K中发生改变。这些可能与预后信息相关。因此,时序分析(timing analyses)已绘制出癌症中不同的进化轨迹或优先的突变序列,这表明肿瘤进化上优化的路径和肿瘤基因突变之间的相互依赖性。
虽然亚克隆驱动因素的存在预示着癌症患者的临床预后较差,但在诊断之前已积累多年的早期克隆驱动因素可能为早期干预提供了机会。换句话说,在患者从病理上被诊断前,大量的早期突变和驱动事件已经发生,如果能“揪出”这些凶手,就为医生及时干预提供了机会。
但是诸如WES和WGS等对整个组织进行Bulk测序,在低CCF条件下有诸多瓶颈,限制了其解决克隆系统发育关系的能力(limited in their abilities to resolve the phylogenetic relationships of clones)。
2.2 多重采样追踪定义克隆动态变化(Defining clonal dynamics through multi-sampling)

癌症进化是一个动态过程,面对诸多挑战,例如制定有效的用药和治疗方案等。适应性不同的克隆亚型,可能会重塑肿瘤的基因组信息。由于CCF随时间变化的协调模式,在克隆进化过程中的不同时间点进行多次采样甚至可以为CCF较低的亚克隆提供更高分辨率的系统发生关系。有了更多的采样时间点,可以在通过CCF识别出与其他亚克隆明显不同的单个亚克隆,尤其是当它们具有明显的生长动态时。此外,serial sequencing不仅可以提高克隆分解(clonal decomposition)的能力,还可以评估克隆特异性和适应性。
实际上,对CLL细胞或实体瘤患者血浆中的ctDNA进行时间点上的密集采样,可以通过一些数学模型和生信算法来评估肿瘤中克隆生长进化动力学(clonal growth kinetics)与治疗之间的关系。例如与治疗前相比,化疗后复发的CLL中TP53突变CCF比例有显著提高。这表明面对治疗这种压力选择时,包含TP53突变的亚克隆适应能力更强。相反,专门针对CLL的靶向治疗方案会使得携带BTK突变的克隆增加,变得更加容易。在结直肠癌中,可以追踪接受EGFR抑制剂治疗患者血浆的ctDNA,来确定耐药性克隆在治疗前后发生的变化。
值得注意的是,克隆生长的数学模型(mathematical modelling of clonal growth)能够进行正推和反推,以预测未来的克隆比例并预估治疗开始时耐药细胞的数量。这些数据还为基于对不同克隆的治疗特异性适应性效应的连续测量,为多种治疗方案的组合而开发的算法,在算法优化上铺平了道路。
多重采样的另一种形式是研究肿瘤内的多个区域,以评估肿瘤内克隆的空间组成。与多个时间点密集采样比较相似的是,非小细胞肺癌NSCLC的多病灶部位采样有助于研究人员更加了解这些克隆之间的关系,从而有助于改善临床上进行分级和诊断。在进化选择的一个显著的例子中,医生对多区域多病灶(如肝癌原发灶和转移灶的多个结节进行采集)采样揭示了驱动拷贝数变异(driver CNA)的趋同进化,因此同一驱动CNA涉及整个肿瘤中不同的亲本等位基因。多区域多病灶采样还可以比较原发灶,与原位复发、复发性转移灶、淋巴结转移等之间的关系,并确定转移灶中的驱动基因。例如,肺腺癌出现脑转移候,脑部病灶中MYC,YAP1和MMP13中的CNA与肺癌结节原发灶之间可以进行比较。
尽管全基因组层面的测序数据如WGS和WES等手段,可以从广度上可以帮助研究人员推断出追溯谱系(retrospective lineage tracing),但微卫星在错配修复(MMR)缺陷的结直肠癌中的高变异性是另一种被广泛使用的标志物,简称微卫星不稳定性((Microsatellite Instability, MSI)。2020年NCCN指南中已经明确将MSI作为结直肠癌预后因子。
· 知识拓展 ·
微卫星不稳定MSI:
微卫星不稳定是由于肿瘤细胞基因组不稳定造成的一种特殊突变现象。微卫星序列指的是基因组上广泛存在的短串联重复,重复单元通常为1-5个碱基。在基因组不稳定的情况下,微卫星位点在复制过程中出现滑动错配,从而出现频繁的拷贝数单元增加或者丢失,形成微卫星位点上的插入或缺失,这种情况称作微卫星不稳定现象。
基因组层面上的微卫星位点,早已被人们视为分子钟,为克隆进化提供了重要的线索。 例如,结直肠癌发生前的大肠腺瘤的分子年龄(molecular age)与腺癌相当,这是腺瘤到腺癌逐步发展的过程。
在最近的一项研究中,20个poly-G重复的微卫星位点在多区域多病灶分析中用于谱系追踪,挑战了“顺序进展模型”(sequential progression model)。该模型假定先发生淋巴结转移,再通过淋巴结转移到远端器官。这项研究表明,在三分之二的病例中,淋巴结转移和远处转移均直接源于原发性结直肠腺癌,对解释活检样品的分期具有明显的意义。
在此经验的基础上,针对微卫星位点采取了类似的方法,以证明在远处转移中,多克隆播种/接种(polyclonal seeding)在淋巴结转移中更为频繁。因此,对转移性恶性肿瘤的分析可以进一步阐明克隆传播的模式,并可以推断出肿瘤进化的迁移模式。
· 知识拓展 ·
克隆播种:
在肿瘤转移过程中,癌细胞从原发灶迁移到转移灶的过程称作播种(seeding)。播种分为单克隆播种和多克隆播种,其中多克隆播种指的是携带不同突变的细胞共同迁移到了转移病灶,导致转移病灶天然地具有瘤内异质性和亚克隆结构。而通常来说,药物作用等强大的选择压力会使得播种过程更倾向于单克隆,并且播种的克隆可能携带新的驱动突变和耐药突变。
2.3 单细胞水平的遗传史追踪(Tracing genetic history in single cells)
尽管多样本/多重采样为解构癌症进化的克隆型提供了较高的分辨率,但仍需要从单个细胞水平的分辨率解析系统发育,才能得出精确的克隆动力学和肿瘤的进化史。通过光学(免疫荧光FISH探针等手段)或单个细胞的条形码高通量技术(以10X Genomics和BD为首的商业化单细胞测序平台)进行的预期谱系追踪,已经在技术上实现了在体内(in vivo)和体外(in vitro)对肿瘤进化建模,包括能够捕获其他细胞特征的方法,如单个细胞的mRNA表达。
但是,上面提到的这些方法不适用于primary human tissues的回顾性谱系追踪,而追溯这些“native barcodes”是关键。
基于Droplet microfluidics微流控单细胞捕获平台,可以捕获上万个细胞的基因型信息(somatic genotypes)。对复发AML的驱动突变进行单细胞靶向DNA测序(single-cell-targeted DNA sequencing),以揭示急性期AML从诊断到复发的亚克隆转变(subclonal shifts)。
直接应用于人类癌症的回顾性谱系追踪推断的另一种方法是单细胞WGS(scWGS)。scWGS可以推断出用作天然条形码(natural barcodes)的CNA,通过该条形码可以绘制单个细胞之间的系统发育关系。一项发表在2011年的乳腺癌单细胞测序研究阐释了乳腺癌肿瘤组织中存在不同的克隆,并证明转移灶是从单个克隆开始向人体其他部位进行扩增和接种的。具有肿瘤非复发特性的复杂CNA的单个细胞(Individual cells with non-recurrent complex CNAs),为研究人员提供了新视角,来研究促进克隆进化的潜在遗传多样性。
近期的一些研究已经引入了高通量scWGS方法,可以对数千个乳腺癌细胞进行测序,并可以鉴定早期驱动基因(例如MCL1和MYC等,与亚克隆的扩增(例如RAD18)。
scWGS的特征在于SNV低覆盖率,数量相对较少的somatic CNA限制了谱系重建的分辨率。此外如上所述,在趋同进化的例子中,在单个肿瘤的两个等位基因中都可以看到相同的CNA,因此请注意不要使用无限位点假设进行经典的系统进化重建(也就是说,用于谱系追踪的突变不会回复突变,不会重复突变,新突变总是发生在之前未突变的位点上)。因此,具有“有限位点”模型的系统发育方法可能更适合解决非整倍体肿瘤。其他技术手段上的限制还包括对单细胞水平的基因组DNA进行全基因组扩增中引入的一些错误,如等位基因缺失,覆盖范围不均匀以及PCR错误等。目前一些课题组已经开发了新的生信工具,可以将经典的系统进化方法用于物种进化,以解决单细胞DNA测序数据中产生的噪音和错误信息。
通过对单个细胞来源的克隆(single-cell-derived colonies)来增加DNA Input起始量,可以消除单个细胞全基因组DNA扩增所产生的错误。换句话说,增加同一个或同一类细胞的数量,就能增加同一细胞来源的DNA,以减少DNA扩增时产生的错误。
对单个细胞克隆(single-cell colony)或类器官数据的高分辨率谱系重建,已应用于正常的造血发育组织和癌组织研究。高分辨率发育树(high-resolution trees)为一些关键推论铺平了道路。这些推论直接源自患者体细胞进化的一些关键参数信息,例如确定正常成年人自我更新的造血干细胞池(self-renewing haematopoietic stem cell pool)的大小和增长率(rate of growth)。这项研究还开始将克隆动力学与谱系命运联系起来,表明人类成年造血干细胞在140个已经被测序的单细胞克隆中贡献了髓样和B细胞(myeloid and B cells),但没有贡献T细胞。虽然该实验的通量受到限制,但它提供了一个模型,用于追溯人类原始样本中正常细胞和癌症干细胞的遗传谱系历史以及命运决定的相关因素,并将其和转录组数据、表观遗传数据等其他组学中的基因突变信息相结合。
03
肿瘤进化过程中细胞状态的异质性
Heterogeneity of cell states in tumour evolution
3.1 细胞状态可塑性作为癌症进化的媒介
肿瘤内的遗传变异性是肿瘤异质性的基础,并提供了可遗传的多样性,可驱动整个癌症的克隆进化。然而,仅遗传机制可能无法获取完整的肿瘤内异质性的相关信息。长期以来研究人员就发现,这种变异性是由于单个肿瘤内细胞状态的可塑性造成的。一个典型的例子是上皮组织类的恶性肿瘤中的上皮-间质转化(EMT),这让人联想到胚胎发育过程中上皮细胞和间充质细胞之间的转化。
· 知识拓展 ·
上皮-间质转化EMT:
EMT(Epithelial-Mesenchymal Transition)指的是上皮细胞转化成具备间质表型的细胞的过程,这个过程广泛地发生于胚胎发育、损伤修复和肿瘤恶变的过程当中。在肿瘤恶变过程中,上皮细胞通过EMT过程获得了迁移能力,从而能够从基底膜上脱出,转移到其他不同部位。
EMT是一种具备差异性的动态变化过程(a heterogeneous and dynamic disposition with intermediary or partial EMT meta-states),它由复杂的转录因子网络进行调控,如SNAI1、SNAI2、ZEB1、TWIST1等。 在临床上,EMT与不同程度的浸润、侵袭和转移潜能(invasive and metastatic potential)相关。
更广泛来说,如同在白血病、脑癌以及上皮肿瘤中所观察到的那样,癌症通常重现了正常的生理发育程序(physiological developmental programmes),因此可能由干细胞样和分化的细胞类型(stem-like and differentiated cell types)组成。这些观察到的结果继而能够支持产生癌症的分级/分层/分化模型(hierarchical model)。
尽管在癌症研究领域仍存在争议,但在该模型中,肿瘤干细胞处于肿瘤分化最高等级。肿瘤干细胞与正常干细胞一样,都有广泛的增殖和自我更新能力(extensive proliferative and self-renewal capacity)。例如,未成熟型急性髓细胞白血病细胞显示强大的繁殖能力,这种繁殖能力几乎与白血病各种亚型都没有任何关系,几乎可以忽视各种突变驱动基因。
与癌症始于具有干细胞样特性的细胞中的结论相一致,干细胞样程序的启动已被一些研究证明发生在恶性转化(malignant transformation)之前。具体来说,在BRAF突变和TP53突变的黑色素细胞(melanocyte)中激活胚胎神经嵴祖细胞状态会诱导黑色素瘤的发生。这些结果表明,体细胞突变与干细胞样两者之间协同作用会引发癌症。
尽管这些研究为干样细胞的靶向治疗提供了令人信服的证据,但肿瘤细胞的可塑性可能会使这一策略复杂化。分化的癌细胞表现出去分化成干细胞样的能力,使得恶性肿瘤细胞同时具备了分化和去分化干细胞样,这两种状态的双重切换能力的可塑性。
细胞状态的随机转变也可以充当癌症抗性的媒介。通过对小鼠或体外培养的细胞进行药物处理,能够鉴定出少量因药物处理而存活的细胞并监控获取这些“稀有细胞”持续性的转录状态。这种状态因为药物等外界选择压力的作用下获得了驱动基因突变继而对药物产生耐药性。
例如将前列腺癌或肺腺癌转化为小细胞癌,或通过Richter转化将CLL转化为弥漫性大B细胞淋巴瘤,这种谱系可塑性(lineage plasticity)一直被认为是生产耐药性的非遗传机制。这种形式的谱系可塑性通过对细胞重编程提供了一种耐药性的新途径,从而消除了对治疗靶向途径的依赖性。
总的来说这些证据表明,癌症进化是遗传多样性与癌症在细胞状态之间进行切换的能力之间复杂相互作用的结果,这使得癌细胞群能够迅速找到适合自己的最优进化轨迹和路线图,从而能够逃脱药物干预和其他治疗方案对其的束缚。因此,需要将细胞状态异质性与遗传癌症进化模型这两种方法整合在一起,解决这个难题。
3.2 单细胞分辨率下的转录异质状态(Heterogeneous transcriptional states at single-cell resolution)
由于转录状态异质性是肿瘤发生和进展的关键因素,因此原发性肿瘤的单个细胞RNA测序(scRNA-seq)已成为转化医学中常用的一种工具或手段。大量的泛癌/跨癌种研究表明,肿瘤内细胞状态异质性已经成为一个约定俗成的准则和“共识”,不再是一种“意外发现”。细胞状态的异质性不仅跟基本细胞过程相关(例如细胞周期,代谢和缺氧诱导的应激状态),还跟发育跟耐药相关。
通过scRNA-seq技术对临床肿瘤样本进行测序,绘制了干细胞样和不同发育层次结构的高分辨率单细胞图谱。例如在IDH1/2突变的少突胶质细胞瘤中,scRNA-seq鉴定出具有神经干细胞程序激活的干细胞样癌细胞,该程序位于层次结构的顶部,分支成两个不同的细胞状态,类似于星形胶质细胞和少突胶质细胞。
与癌症干细胞模型一致,细胞周期相关基因的表达在干样细胞中高度富集。在黑色素瘤的单细胞测序结果中,干性信号与对RAF和MEK抑制剂的AXL高耐药性程序相吻合,将关键特性(例如生长和对治疗的耐药性)直接对应到了同一细胞上。
scRNA-seq能够进一步检测出治疗后患者中的仍然驻留稀有癌细胞。基于治疗性骨髓瘤的已知恶性特征,对多发性骨髓瘤的治疗前和治疗后的scRNA-seq数据进行比较后,可鉴定出残留的肿瘤细胞(通常占浆细胞群的一小部分)。
因此,scRNA-seq可以区分非肿瘤细胞中的稀有肿瘤细胞,以对疾病进行早期筛查,或鉴定出残留的少量癌细胞并迅速锚定可能会产生耐药的细胞。
此外,scRNA-seq不仅可以在既定的时间范围内测量细胞状态,而且新开发的生信工具还可以利用scRNA-seq数据来模拟细胞状态的动态变化轨迹。基于转录数据中相似性梯度的推论,已经出现了各种算法来重构细胞的分化轨迹,这种分析被称作是拟时序分析或伪时序分析pseudotime projection analyses。
例如R包Monocle能够生成构造了最小的生成树(spanning trees),将单个细胞mapping到主干上。常规的细胞生物学研究要分离特定的细胞,需要针对该细胞的表面蛋白定制特异性抗体。该算法的优势在于,scRNA-seq数据中的细胞本身不依赖于基于已知细胞表面marker蛋白,因此可以捕获细胞的一些过渡状态,有助于研究人员判断细胞状态动态变化。
另一种算法则通过scRNA-seq中未剪接的mRNA与已剪接的mRNA的比例来确定RNA速率,即RNA velocity。通过mRNA丰度的变化率,来预测未来的细胞状态。由于“打开”的基因比“关闭”的基因具有更高的比率,因此RNA速率可以预测细胞所假定的转录谱,从而可以预测未来的细胞状态并测量当前的细胞状态。
这两种方法都能够对正常组织和癌组织中细胞状态动态进行监测。值得注意的是,这些方法与遗传谱系追踪无关,因此通过单细胞多组学整合将细胞状态动态与遗传同一性联系起来,这个前景相当诱人。从而阐明了体进化中细胞状态动态的遗传基础。
3.3 遗传谱系历史与单细胞转录状态关联(Coupling genetic lineage history with single-cell transcriptional states)

用不同的表型变异(例如沿着developmental hierarchies或治疗后产生的耐药性)来追踪癌细胞的谱系历史,需要将遗传信息与scRNA-seq数据直接整合。例如通过将患者的病灶移植到异源物种如小鼠体内如PDX等,这些移植的组织或细胞用慢病毒转入了一些条形码信息。具有相同条形码(即源自同一细胞)的肿瘤细胞占据不同的谱系特异性状态,从而证实了胶质母细胞瘤细胞的可塑性。但是这种工具不能用于primary human tissue,而需要用到体细胞突变基因条形码信息。
目前已经有一些课题组开发了在单个细胞中整合DNA和RNA表达的实验方法。例如,G&T-seq可以通过提取和分离基因组DNA和mRNA进行扩增和测序来捕获单细胞基因组信息和所有mRNA的转录组数据。另一种方法sci-L3使用组合index提高了双重测定的通量。
如上所述,这些技术具有跟scWGS的相同局限性,例如测序数据和算法可拓展性差(scalability),测序数据本身的稀疏性(sparsity)、PCR错误(PCR errors)和等位基因缺失(allelic dropouts)。scWGS的应用对于大部分整倍体恶性肿瘤的所能提供的帮助比较有限,因此靶向测序可能会提供更多信息。尽管如此,这些方法可以将遗传谱系历史与同一细胞中的非整倍体肿瘤的表型状态联系起来。
· 知识拓展 ·
1. scalability
scalability字面意思上解释为可拓展性,实际上在高通量测序以及计算机科学里面,scalability泛指那些计算能力进行弹性拓展和算力提升的一种状态。Scalability is the property of a system to handle a growing amount of work by adding resources to the system。但是在本文负面语境中特指lack of scalability的意思,意思是可拓展性差。
2. sparsity
sparsity字面意思上可能直接可以翻译成稀疏性,在人工智能领域泛指那些需要处理的数据稀疏化,如降低复杂度正则化。或者部分神经网络里面的参数过于复杂,需要对模型中过于复杂的参数进行简化达到模型稀疏性的目的。这就导致在单细胞测序领域这个解释有所不同。只要测的细胞较多,而每个细胞的转录本又较多,这个现象总会存在。在单细胞中通常和dropout联系在一起,意思就是测不准。在表示转录本丰度的矩阵中,对象和属性都很多,每增加一个只有少量属性值的对象,就会带来大量的零值。
此外,通过评估跨染色体的基因表达的不平衡,转录组本身丰富遗传信息已被用来推断CNA的大量扩增和丢失。在这些非整倍体癌症 的早期标志性单细胞研究中,这种方法使得能够研究克隆与发育状态之间的联系。
在少突胶质细胞瘤中,尽管可以在不同的细胞状态中鉴定出亚克隆的倾斜富集,但克隆差异与主要细胞状态不符。在胶质母细胞瘤中,特定拷贝数的增加(例如分别在EGFR,CDK4和PDGFRA中的拷贝数增加)促使肿瘤细胞向星形细胞、神经祖细胞和少突胶质祖细胞状态发生转换。
此后一些改进的生信算法已经整合了用于复制中性杂合性丢失(copy-neutral loss-of-heterozygosity events)的SNP,以及CNA的整体基因表达水平。这些方法在分析多发性骨髓瘤中,将初始癌细胞中的亚克隆缺失del16与类似于复发性髓外骨髓瘤细胞的转录特征联系起来,表明肿瘤的侵袭性与原始肿瘤中亚克隆驱动因子之间的潜在联系。
通过Smart-seq2 protocol,将SNV驱动基因信息与scRNA-seq数据进行整合,可获取全长转录本。已知在34%的肿瘤细胞中促癌组蛋白H3K27M中都发生了突变。得益于Smart-seq2的polyA扩增方式,与未聚腺苷酸化(non-polyadenylated)的HIST1H3B相比,聚腺苷酸化(polyadenylated)的转录本H3F3A的效率更高。
接下来通过在Smart-seq2原有的protocol中添加H3F3A突变基因座特异性引物(44个分析细胞中的97%),显着改善了检测效果。目前已经开发出与Smart-seq2的类似方法,将BCR-ABL阳性CML干细胞的基因改变和转录状态联系起来,以追踪和揭示TKI抑制剂治疗效果。
总而言之,这些研究表明,克隆差异与细胞状态异质性保持着复杂的关系,不同类型的肿瘤之间存在差异。进一步表明需要以更高的通量将遗传信息与单细胞水平的转录状态进行整合。
3.4 单细胞转录组中的体细胞基因分型(Somatic genotyping in high-throughput single-cell RNA-seq)
由于Smart-seq2较低的通量和reads覆盖较低,上述研究中的体细胞基因分型效率很低。而通过对基因组上的DNA和cDNA进行靶向扩增的高灵敏度的体细胞基因分型,能够在慢性髓样肿瘤(chronic myeloid neoplasms)整合克隆结构和转录状态。
基因组DNA扩增的另一个优势是对低表达或未表达突变的基因分型进行鉴定。尽管此方法是在Smart-seq2 protocol基础上改进的,但通过开发384孔板可生成3'转录组文库,其通量由原来Smart-seq2的1个细胞提高到了数千个细胞。每个细胞最低30,000到50,000个reads,部分文献每个细胞达到了0.2M 个reads。
10X Genomics的单细胞转录组也推出了SNV的检测,每个细胞至少大于20,000个reads。这种方法提供了一个框架来解析AML的亚克隆转录身份,例如GATA2突变的亚克隆。
本综述作者和其他课题组对传统的基于纳米孔scRNA-seq平台进行了改进,通过靶向扩增感兴趣的突变基因barcode cDNA,在成千上万的单细胞中与全转录水平的mRNA一起,能够实现高灵敏度的体细胞突变信息捕获。

van Galen等人利用基于纳米孔的scRNA-seq技术,在AML中实现了靶向的体细胞基因分型鉴定。这些数据已经证明,反映造血分化状态的AML转录身份与潜在的遗传驱动因素密切相关,与前面提到的胶质母细胞瘤的基因型和细胞状态非常相似。
例如,在原始祖细胞状态中,FLT3内部串联重复(internal tandem duplications)与在同一肿瘤内分化细胞中的FLT3 Asn841Lys突变紧密相关。
尽管纳米孔的通量能够实现数千个细胞的转录组分析,但由于目标基因的表达水平较低,且目标基因位置与转录本末端距离较远(例如10X Chromium只能捕获3’端的部分mRNA信息),大多数目标基因突变位点捕获的效率较低。但是,同时捕获全转录组信息可以通过随机森林分类器(Random forest classifier),基于基因型细胞和非基因型细胞之间的转录相似性推断突变状态。
作为一种替代方法,本文作者基于10X Genomics平台开发了转录组基因分型(GoT)技术。GoT能够在数千个细胞中进行高度敏感的基因分型鉴定,靶向CALR驱动基因中主要的体细胞突变基因分型效率约为90%。
尽管如此,由于10X技术本身的限制导致无法获取全长mRNA,这就使得目标基因区域与3’末端之间的距离限制了某些目标基因分型的鉴定。为了克服这一局限性,作者引入了两种补救的方法,包括long reads测序和GoT环化建库,其中连续几轮的环化和反向PCR,能够去除靶向区域序列和细胞barcode序列之间的插入序列,换句话说这2个补救方法能够把不必要的序列给去除,从而产生了可以用于标准分析流程的短reads测序。

作者还注意到,由于对scRNA-seq中的对mRNA pooling进行扩增时容易产生偏好性,如上图所示,由于PCR扩增不仅容易产生非线性扩增还容易在PCR过程中引入错误,这就是导致对突变信息可能被错误地指定为野生型。于是作者引入了UMI分子标签。作者还系统地检查了基于基因型的发现与改变最小UMI计数阈值之间的关系,以评估结果的稳健性。虽然GoT在检测低表达基因上仍然面临诸多的挑战,但对scRNA-seq平台优化仍在继续,提高总体RNA捕获效率将提高基于cDNA的基因分型鉴定的概率。

GoT在高通量下共同捕获细胞状态和基因型信息的能力表明,在CALR突变的骨髓增生性肿瘤中,造血祖细胞的主要转录信息与体细胞突变状态不相关。也就是说,突变细胞和野生型细胞混合在整个造血分化树中,这证实了精确的基因分型对于区分突变体和野生型细胞的重要性。
因此,在这种情况下,GoT能够将突变分化轨迹叠加到同一个体内造血发育的正常图谱上,从而消除了潜在的生物学混杂因素和技术批次效应。对突变和野生型分化细胞图谱的直接比较发现,突变细胞的频率随着髓样分化的增加而增加,表明差异适应性优势是祖先身份的功能。
为了证实这一结果,作者将拟时序分析与细胞的遗传图谱进行了整合,并证明与野生型细胞相比,突变的细胞偏向分化状态。与造血干细胞相比,在分化的祖细胞如巨核细胞祖细胞中突变相关的细胞周期基因表达增加更为明显。突变诱导的未折叠蛋白应答,NF-κB活性和JAK-STAT信号转导均随祖细胞类型和成熟阶段而变化,表明潜在的细胞特性限制了驱动突变的影响以及由此产生的癌症细胞表型是细胞身份与体细胞突变之间相互作用的功能。
这种方法在研究人组织中的早期克隆扩增中可能特别有用。最近,在全身都发现了形态正常组织内的克隆扩增,导致了体细胞嵌合体现象(mosaicisms)。在环境压力下皮肤、食道和肺等上皮细胞比较丰富的组织中,这些克隆通常在已知的一些驱动基因如TP53和NOTCH1中带有体细胞突变。
类似地在造血系统中,经常在正常的个体中以低等位基因频率证明骨髓恶性肿瘤的复发性驱动基因突变,例如DNMT3A和TET2突变。这种状态被称为“克隆性造血功能”,但使这些人更容易患上血液癌和心血管疾病。然而,对于这些突变的克隆表型产生的表型后果,仍然存在一个关键问题。
因此,将多组学技术应用于这些其他可能正常的组织,有助于识别最早期克隆异常生长的信息。
将高灵敏度的体细胞基因分型鉴定技术,扩展到更多的基因位点还可以实现对克隆的重建,高分辨率追溯谱系追踪(high-resolution retrospective lineage tracing)并与细胞身份和细胞状态信息相整合。
为此,任何突变信息,如线粒体DNA或微卫星位点中的突变,都可以用于谱系标记。将这些天然存在的遗传条形码信息整合到多种单细胞测序的组学技术中,可以提供高分辨率的系统发育数据和细胞发生细微状态变化的数据,最终使我们能够破译癌细胞命运的密码。
04
癌症进化中表观遗传的可塑性
Epigenetic plasticity in cancer evolution
4.1 表观遗传学的基础是细胞状态(Epigenetic profiles underlie cell states)
前文提到的致癌表型,例如持久性(persistence )、自我更新和谱系可塑性等,需要不断遗传给后面的细胞,才能促进癌症向进展和耐药方向演变。但是,这些细胞状态经常被证明在遗传性存在不一致的情况。越来越多的数据表明,细胞状态可以在表观遗传层面进行编码和繁殖,这与正常发育生物学中细胞身份的表观遗传编码(epigenetic encoding)是一致的。 表观遗传学包括DNA甲基化、染色质开放程度和组蛋白修饰,主要通过关键转录因子来激活介导。总体而言,表观遗传修饰引起转录活性的高度协调调节,这对于组织的正常发育至关重要。
血液肿瘤中表观遗传修饰基因(如TET2、DNMT3A和ASXL1)和实体肿瘤中SWI/SNF(BAF)染色质复合体,普遍存在大量突变,这表明表观遗传学在介导肿瘤发生中具有重要意义。另外,一些生化机制研究揭示了表观遗传学在persistence和发生耐药的潜在转录特征。
在胶质母细胞瘤中,肿瘤干细胞显示出H3K27me3的整体重构,并且与Notch信号传导和静默相关的用于激活增强子的组蛋白H3K27ac在部分病灶中表达增加。在雌激素受体阳性(ER positive)乳腺癌中,KDM5组蛋白脱甲基酶的表达增强了转录异质性和耐药性。
因此,越来越多的证据表明表观遗传编码对癌症细胞状态的重要性。
4.2 表观遗传崩塌激活肿瘤可塑性(Corrupted epigenetic identities enable tumour plasticity)
从第一个转化的细胞忠实地传播表观遗传身份,从而可以推断出起源细胞类似于祖先癌细胞遗传信息的忠实传播。尽管如此,表观基因组在不断增长的恶性癌细胞群体中也在多样化信息。因此,正如基因组中的随机突变导致遗传性肿瘤的异质性一样,表观基因组中的随机突变会产生表观遗传的瘤内多样性。这种结果是基于前期大量的DNA甲基化研究得出的。这些研究表明,成千上万个碱基位点表现出“嘈杂”随机模式,代表了正常细胞和癌细胞表观基因组中可遗传的巨大差异。
在这些研究的基础上,对原发癌组织的检查表明,肿瘤细胞与基因进化都表现出DNA甲基化水平的多样化。对前列腺腺癌的原发灶、转移灶和癌前病变的多个组织区域搜集样本,通过DNA甲基化异质性进行的谱系追踪密切反映了基于拷贝数遗传多样性建立的系统发育关系,并构建了进化树。
同样,在结直肠癌模型中,对单细胞来源的结肠癌类器官的DNA甲基化分析显示,与遗传多样性相似,DNA甲基化异质性能够稳定传播。大规模CLL队列样本DNA甲基化测序结果证实,肿瘤内DNA甲基化的异质性高于正常B细胞。
多个相互协调的表观遗传层,能够调节基因表达和细胞身份,早期数据表明表观遗传多样化超出了DNA甲基化的水平。例如,表观基因组各层之间的协调遭到破坏,导致共同mapping到相同的基因组位点,后者通常在CLL中专门激活和抑制组蛋白修饰,这可能反映了组蛋白修饰中的细胞间多样性,并与更大的转录多样性相关。

重要的是,最近的证据支持这样一种观点,即选择可能对表观遗传多样性起作用,类似于公认的克隆进化遗传多样性模型。在IDH突变型神经胶质瘤中,CTCF绝缘子蛋白结合motif的随机超甲基化导致增强子和基因之间的绝缘(insulation)丧失,这可能被强行控制为致癌基因激活的机制。
相反,通过启动子高甲基化或多梳蛋白polycomb介导的抑制,异常限制性状态可以抑制分化程序的诱导,从而将癌细胞停滞在增生状态,如EZH2突变的B细胞淋巴瘤所示。最后,通过表观突变的过程破坏了表观遗传编码(例如,随机DNA甲基化变化)可以降低细胞状态之间转换的障碍。这种现象可能是恶性肿瘤可塑性增强,破坏分化等级并促进诸如去分化为干细胞样等过程的基础。这种可塑性分化拓扑结构已经通过数学模型显示出来,可以放大阳性选择,从而扩大癌症的进化能力。
综上所述,表观遗传信息作为关键细胞状态的重要遗传信息而出现,因此可以为选择和进化提供额外的作用机制。
4.3 多组学技术整合单细胞遗传/表观/转录信息(Multi-omics technology links genetic, epigenetic and transcriptional information in single cells)
目前全球各地实验室正在开发新的生信算法和新的实验方法来捕获单细胞中的表观信息和细胞状态信息,如同一细胞进行蛋白和转录信息的捕获。整合了蛋白质表达的scRNA-seq和单核染色质开放程度检测(ATAC-seq)已在同一癌症样品上并行进行测序,根据染色质开放程度推断分析了转录状态与调控网络失调之间的联系。
在混合谱系双表型白血病(在同一白血病中具有髓样和淋巴样分化的标志物)中,将肿瘤细胞mapping到具有mRNA和ATAC-seq数据的正常造血分化图可以将转录因子激活与其下游靶点建立因果联系,如RUNX1上调CD69。此类分析或实验结合的双高通量scRNA-seq和ATAC-seq方案(如sci-CAR)的进一步应用,可能有助于识别精确的顺式调控位点和靶基因,并确定关键的调控网络控制克隆进化。
基于Smart-seq2 protocol基础上进行修改后,可以对同一细胞来源的DNA甲基化信息和mRNA转录组信息进行同时捕获。一种方法是将scTrio-seq用于肝癌和结直肠癌的多区域采样,以在转录和甲基化状态的背景下追溯癌细胞的遗传谱系历史。在结直肠癌中,甲基化图谱与CNA定义的遗传谱系紧密相关。尽管转录程序与启动子的甲基化呈负相关,但正如预期的那样,转录状态与亚克隆遗传特性并不一致。
将DNA甲基化和整个转录组数据链接到单个CLL细胞中,发现细胞间甲基化变异与基因表达变化密切相关。因为每个细胞捕获约10%的目标甲基化组的捕获率,尽管单细胞基因组数据中稀疏性的局限性,在单细胞亚硫酸氢盐测序数据中也遇到了相应的瓶颈,但DNA甲基化的整体变化为克隆进化等提供了宝贵的见解,例如测量表观层面变异的能力。值得注意的是,可遗传的DNA甲基化突变也可以作为分子钟,因此可以用作天然条形码,直接推断原发性CLL样品中肿瘤细胞的高分辨率系统发育史。
体细胞基因分型在此多组学方法中的进一步整合应用于亚克隆SF3B1突变,这表明突变的细胞聚集在基于DNA甲基化的系统发育树的一个进化枝中,具有不同的转录输出,为树推断提供了正交验证,并能够进行估计SF3B1突变获得时的淋巴结年龄。
最终,转录信息直接在谱系树上的多组学投影揭示了暴露于不同进化枝中不同途径的治疗性暴露。尽管联合单细胞表观遗传多组学平台在癌症上的应用仍处于起步阶段,但这些数据突显了将各种可遗传但可塑性的癌细胞单细胞整合用于跟踪(并最终预测)克隆进化的希望。
05
细胞空间动态与肿瘤微环境
Spatial dynamics and the microenvironment
5.1 细胞空间动态是肿瘤异质性的遗传来源(Spatial dynamics as a heritable source of tumour heterogeneity)
细胞空间定位代表与适应性相关(associated with fitness)的肿瘤细胞进化中的另一个可遗传的维度(heritable dimension),因为肿瘤细胞倾向于与其亲本细胞(parent cells)定位在一起,并且其生长程度受到了一定限制。
例如,在某些情况下,实体瘤中边缘的细胞会处于一个活跃的细胞周期(“边界约束”生长boundary-bound’ growth),这表明空间限制是与转移潜能和生存相关的重要的可遗传性特征。
最新数据表明,肿瘤细胞的空间分布不仅与适应性(fitness)有关,而且与克隆进化有关。与同一尺寸的恶性前腺瘤形成鲜明对比的是,同一肿瘤内单个腺体(由<10,000个细胞组成)的多区域多部位采样结果,进一步显示了结直肠癌中的多种克隆高度混合,这证明了克隆身份的分离(segregation of clonal identities)。
与这些数据一致,数学模型算法模拟的预测结果,确定了肿瘤细胞的克隆混合是结直肠癌的早期事件。因此,类似于癌细胞的表观遗传学特征的持续崩坏/失控(corruption),组织结构的破坏(breakdown)可能是克隆进化的关键特征,因为即使微小的细胞扩散也会增加肿瘤的适应性及其对当下需要面临关于治疗方案的挑战。
值得注意的是,尽管白血病不受组织在空间上的严格限制,但骨髓本身的混乱无序(disorganization)是髓系肿瘤的长期存在的一个现象和特征。因此,组织结构的畸变是实体瘤和血液恶性肿瘤之间进展的共同特征。
肿瘤进化过程中的空间嵌入(Spatial embedding)还会导致不断增长的恶性癌细胞群体所在肿瘤微环境的相互作用。在所有癌症中,包括免疫细胞,内皮细胞和基质细胞在内的肿瘤生态系统已被证明是癌症转移、进展和对治疗应答的关键决定因素。
近期一项研究揭示了一个令人惊讶的现象,非小细胞肺癌以与特定免疫微环境高度相关的方式,通过启动子甲基化或CNA调节新抗原的表达以进行免疫逃逸 。
同样,在恶性程度较高的晚期卵巢癌中,进行多区域多病灶部位采样后观察到肿瘤与免疫细胞的相互作用,表明与肿瘤相关的T细胞与克隆异质性呈负相关,倾向于选择具有免疫逃逸机制的克隆。
这些研究表明,在相同的恶性癌细胞种群中,在基因组或表观基因组水平上可遗传的进化变化,会随着与非恶性细胞相邻的局部相互作用而变化。具体而言,这些研究表明,肿瘤微环境正在对亚克隆施加选择性压力,并积极促进癌症的进展。
5.2 单细胞水平分辨率的肿瘤微环境(The tumour microenvironment at single-cell resolution)
虽然上述对原发灶肿瘤临床样本的大量多组学分析,为肿瘤微环境对进化过程的影响提供了有力的支持,但肿瘤微环境的细胞复杂性要求单细胞分析以提供其与肿瘤相互作用的高分辨率图。
头颈癌scRNA-seq测序结果中,鉴定了具有部分EMT程序的肿瘤细胞与基质划分/基质间隙(stromal compartment)之间的相互作用。
最近scRNA-seq也证明了肿瘤微环境的复杂性,揭示了大量支持免疫逃逸的免疫微环境偏好性等相关证据,例如通过对转移性黑色素瘤多细胞微环境中分离的T细胞来支持免疫逃逸理论。
实际上,单细胞测序已经在免疫学和免疫肿瘤学领域发生了变革。然而,对免疫细胞生物学的深入讨论超出了本文范畴。
然而,由于scRNA-seq通常需要对新鲜组织进行解离,因此不能保留肿瘤细胞和微环境的结构信息。尽管如此,仍可以通过关联已知配体和受体的表达,从scRNA-seq推断出细胞间的相互作用。
虽然已经有开发了大量分析工具来预测高度结构化的组织结构的空间排列,但高度混乱的癌组织可能对基于解离样品的scRNA-seq进行推论更具挑战性。
5.3 单细胞空间识别/可视化技术检视肿瘤微环境(Examining tumour microenvironments with spatially aware single-cell technologies)
在肿瘤微环境的研究中,具有在空间水平检测单细胞的技术平台可能会产生革命性突破。对于蛋白质检测而言,使用同位素(如质谱细胞成像IMC和离子束成像MIBI图像),或荧光团(如多重免疫荧光MxIF和循环免疫荧光CycIF)进行多重标记可以检测细胞或亚细胞分辨率上的数十种标记。
当这些工具应用于肿瘤临床样本后发现,肿瘤内呈现出显著的异质性,包括与空间位置相对应的信号通路改变。通过免疫荧光技术,如庄小威发明的merFISH单细胞成像技术以及seqFISH等用于检测单个细胞中的mRNA信息。此外利用FISSEQ和STARmap扩增cDNA的原位RNA测序已将目标基因增加到数百个基因。
利用UMI技术检测mRNA分子的空间转录平台极大地拓宽了研究维度,之后将这些数据与高通量scRNA-seq平台进行联合分析,从而可以基于空间模式化的基因表达将来自scRNA-seq数据的分离细胞重新对应到其空间位置。
通过整合目标mRNA分子的原位杂交,从而扩展了IMC平台,同时在单个细胞水平实现了蛋白表达和基因表达,从而可以确定RNA与蛋白质表达的相关性。例如,在乳腺癌中,HER2基因和蛋白质表达高度相关,而CK19显示蛋白质与相应基因水平之间的相关性较弱。相同的方法鉴定了在与T细胞相关的肿瘤细胞中表达的T细胞募集细胞因子CXCL10,从而对肿瘤免疫细胞有了更加深入的了解。
目前还有其他课题组相继开发了许多生信分析工具,以量化关于空间定位和细胞间相互作用的细胞内源性与环境性因素相关的基因表达或蛋白质表达。
这些细胞空间技术平台的应用会与高通量单细胞多组学结合在一起,有望增进人们对细胞间相互作用和空间位置的理解。
06
讨论与展望
Conclusions and perspectives
肿瘤进化包括遗传水平、细胞状态水平、表观遗传水平,空间水平和肿瘤微环境因素的复杂相互作用。 最近,新开发的多组学技术已开始在单细胞水平的分辨率上有所突破,能够整合肿瘤进化上的一些遗传因素和非遗传因素。
这些方法通过对临床样本的研究,为解决有关癌症进化的核心问题铺平了道路。例如,相对于细胞在克隆扩增能力上的退化,需要鉴定出促使细胞出现恶性转化的驱动因素,就要求具有对细胞进行基因型分型鉴定的能力,并搜集到驱动突变引起的转录状态和表观遗传状态的相关变化信息。
恶性肿瘤研究的另一个开放领域是评估肿瘤干细胞的家系命运的决定因素(lineage fate decisions),这些决定因素最终取决于体细胞突变与细胞状态的相互作用。这些细胞状态可能反过来由内在因素(表观遗传)或外在因素(环境因素)影响,强调了单细胞多组学整合分析的重要性。
正常组织(normal tissue)中的恶性肿瘤和克隆扩增的单细胞多组学联合分析,可能有助于揭示潜在的模型。在该模型中,多细胞人宿主(multicellular human host)能够抑制凶猛无序的细胞进化过程。
近期测序数据中,人类正常组织的体细胞驱动因子突变的普遍表明,遗传限制/遗传约束(genetic constraints)可能需要与其他机制协同行动以抑制体细胞进化。所谓的遗传约束指的是多个驱动突变时间累积所需要的时间。这个机制就涉及到细胞在空间上的位置与限制。例如在结直肠的隐窝下,该隐窝通过将结直肠中的干细胞总群分裂为孤立的生境(isolated habitats)而减少了有效群体大小,从而有利于遗传漂移而不是遗传选择。
恶性肿瘤进展加上肿瘤细胞在空间层次上更为复杂的结构,会将选择性扩大并对药物产生耐药性。另一种这样的机制可能是基于复杂的层次结构。这些概念的提出在部分构建的数学模型中得到了验证,肿瘤进化树反映了多样化的分化层次结构,并对人体组织正向选择有抑制作用(suppresses positive selection)。
相反的是,当癌症中DNA甲基化“表观遗传封印”(epigenetic identity barriers)逐渐被解锁后,与其相关的去分化模式可能会起到反作用,从而导致肿瘤进化树上正选择(positive selections)变多。值得注意的是,这些分化层次可以通过外部编程或内部编程的方式来实现。外部编程包括利用细胞因子刺激上皮细胞较多的组织,内部编程包括对骨髓等空间限制不多的组织进行表观层面的操作。
· 知识拓展 ·
正选择(Positive selection):
进化过程中,环境产生的选择压力会对新发的突变产生不同的选择作用,其中正选择指的是新发生的突变倾向于使个体产生更适应环境的性状,生存能力提高,从而携带该突变的个体在群体中的占比上升。在肿瘤的进化上,携带驱动突变的细胞会在此过程中获得更加有助于癌细胞生长生存的特性,从而展现出正选择的过程。
由于克隆产物是突破体细胞进化障碍(barrier)的第一个关键步骤,因此他们倾向于影响上皮组织中与细胞因子相关的机制如Notch与Wnt通路上相关基因突变,以及造血器官中的DNA甲基化修饰等。于是,不断增长的克隆可以作为肿瘤进化过程中非常“优良的底物”,最终导致恶性肿瘤相关克隆胜出,占据主流。
换而言之,设想单细胞多组学技术应用场景拓展到整个人体组织的进化,为人体基本系统特征(basic system properties)提供了诸多关键线索。这些特征阻止了亿万细胞废除自身多细胞契约(multi-cellular contract),以牺牲宿主本身为代价做适应性优化。
这些借助多种技术手段的整合分析可以对体细胞演化进行多层次的解析和探究,从而深入多细胞物种演化和单细胞物种无性生殖之间的交叉领域,在这一令人兴奋的交叉点得到新的重大突破。