


黏多糖贮积症(MPS)是一种具有遗传风险的代谢性疾病。因细胞内的溶酶体异常,导致机体中的某些黏多糖代谢紊乱。细胞的正常功能受到影响,并进一步伤害器官的结构和功能。
MPS并不是猫咪专属的遗传性疾病,人类铲屎官中同样也有一定概率患有类似的MPS。
在人类的MPS中,临床根据致病基因和病症表现的差异,将MPS分为了 Ⅰ,Ⅱ,Ⅲ,Ⅳ,Ⅵ,Ⅶ,Ⅸ型等7种类型。
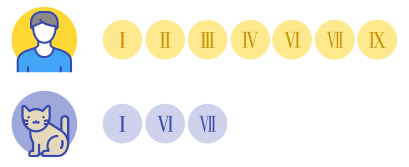
而在猫咪的MPS中,我们暂时发现并归类出三种类型,分别为Ⅰ,Ⅵ,Ⅶ型。

黏多糖(GAGs)又被称作“糖胺聚糖”,是广泛存在于机体细胞中的大分子物质。黏多糖不单单仅指一种物质,它们拥有着一个大家族:硫酸软骨素、硫酸角质素、硫酸皮肤素、透明质酸、硫酸肝素等等...
黏多糖家族时刻为我们的身体提供着强大而又复杂的组合功能。

我们的皮肤是否白嫩,关节是否灵活,心脏是否强壮,眼睛是否明亮...都与黏多糖家族中的每一个成员有着密不可分的关系。
想让“黏多糖”充分发挥价值?首先,身体需要找个地方来处理消化一下它们。
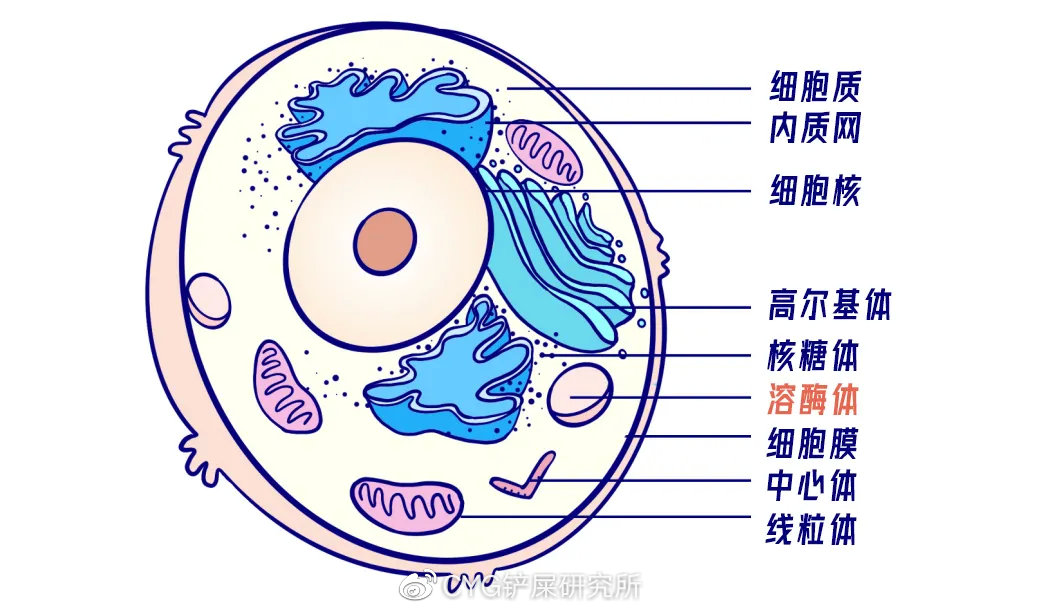
而“溶酶体”便是细胞专门组建的一个「超级处理工厂」。

溶酶体是真核细胞内专门用来降解外来或内源生物大分子的细胞器。
细胞吸收的脂质、糖原和黏多糖等物质,都需要先在这个“工厂”中进行流水线处理。
溶酶体工厂的正常运行离不开工厂中多达60多种的酶。这些复杂种类的酶中“罢工”了任何一个,都可能导致某一特定的生物大分子不能正常降解。
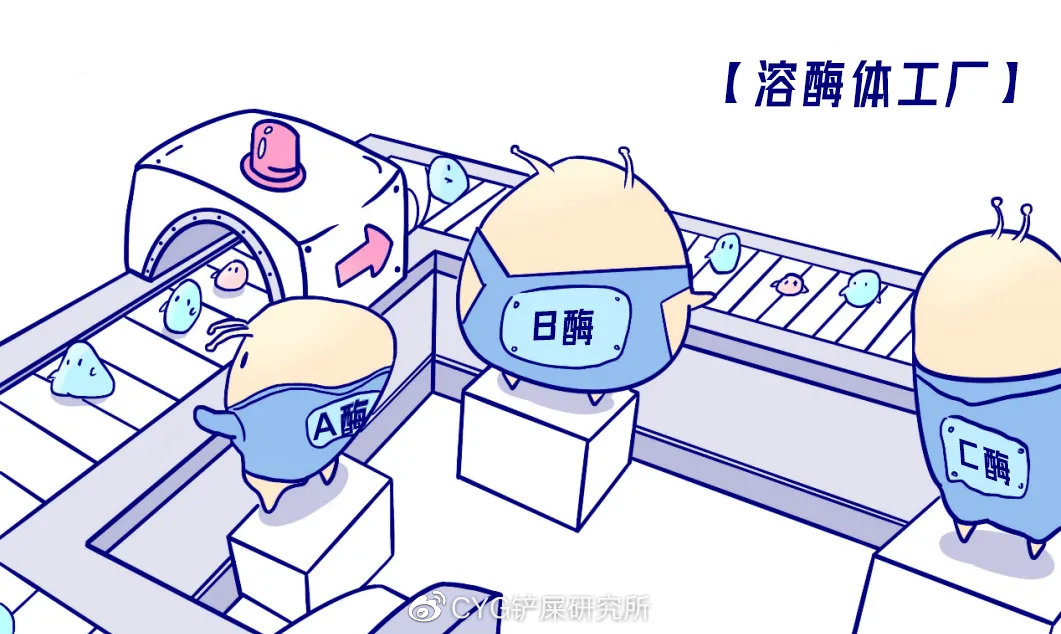
当某种酶“罢工”缺席后,
猫咪的身体会发生什么?
我们刚提到了猫咪的MPS分型。目前,在猫咪身上暂时发现了MPSⅠ,Ⅵ,Ⅶ三种分型,每一种分型都是由不同的基因位点突变而导致,这也最终决定了猫咪体内不同酶的缺陷。
以MPS-Ⅰ型为例:当定位在染色体上22~22q11的相关基因发生突变时,会导致猫咪天生缺乏「α-L-艾杜糖醛酸酶(IDUA)」。IDUA的罢工缺席,让身体没有足够的特定酶来消化分解“硫酸肝素和硫酸皮肤素”。
随着此类黏多糖分解发生障碍并不断累积,溶酶体开始肿胀,最终让细胞也臃肿失常。这些消化不了并不断沉积的大分子让体内各种健康组织细胞逐渐“病倒”,最终引发各种相关病症。

同样地,猫咪MPS-Ⅵ型是由于相关基因突变而导致细胞缺乏「芳基硫酸酯酶B(ARSB)」,而造成组织细胞累积硫酸皮肤素与硫酸软骨素。
猫咪MPS-Ⅶ型是由于相关基因突变而导致细胞缺乏「β-葡萄糖醛酸糖苷酶(GUSB)」,而造成组织细胞累积硫酸肝素与硫酸皮肤素。

目前,猫咪黏多糖贮积症Ⅰ型与Ⅵ型可以通过基因遗传病筛查进行检测。
MPS-Ⅰ与MPS-Ⅳ都为常染色体隐性遗传疾病。当猫咪携带一个致病基因时,可能从外观来看没有任何的异常,但猫咪会有一半的概率把致病基因遗传给下一代。
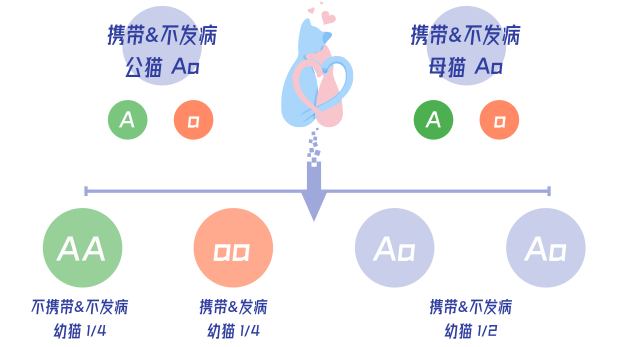
当将两只各携带一个致病基因的猫咪繁育后,幼猫后代中会有3/4的比例携带致病基因,同时也将有1/4的幼猫后代携带纯合致病基因并发病。

在MPS-Ⅰ型患猫体内,几乎每个器官和组织中都会受到病情的影响,主要表现为:内脏器官病变、骨骼畸形和智力障碍。
猫MPS-Ⅵ型和Ⅶ型的类型临床表现与Ⅰ型相似,包括:面部畸形、角膜混浊、肝脾肿大...等等。
在常见的猫咪品种中,暹罗猫、东方短毛猫、巴厘猫、东奇尼猫及混血猫种等...都属于较为高发的品系。

MPS的病况会不同程度地影响患猫的健康福利与寿命。
通常患猫在7岁龄前死亡,轻度患猫会适当地拥有更久的寿命。但无论如何,复杂而又痛苦的病症将会陪伴MPS患猫一生。

在人类的MPS治疗中,会优先采用不同的「酶替代疗法」来帮助MPS各类型患者减轻病情所带来的痛苦和不适,但存在着药费高昂、预后较差等问题。
目前,并没有很好的方法可以完全根治这种疾病。同时非常可惜的是,兽医临床中暂无完整有效的治疗方案来应对猫咪的MPS。
通过基因筛查猫MPS-Ⅰ&Ⅵ,可以及时有效地找到致病基因的携带猫咪。

对于所有携带致病基因的健康猫咪,可优先考虑停止及避免繁育计划,从而长期有效地提升子代健康福利。
对于所有携带致病基因的患猫,可及时发现MPS风险,并在日常的养护中提前进行具有针对性的饲育方法,做好与患猫充分面对疾病的准备,通过科学有效的方式延长爱猫的生命。