

采用建模与模拟探究处方对低溶解度药物吸收的影响—环丙沙星

导读

FDA的研究人员先建立BCS IV类药物环丙沙星的机制性吸收模型,接着通过模型探索环丙沙星体内溶出和各肠段吸收之间的关系;结果表明:小肠下端的渗透性将显著影响环丙沙星的体内行为;药物在小肠上端的总溶解量会影响该制剂的生物利用度。模型还考察了具有体内外相关性的溶出条件;考察导致药物在肠道发生沉淀的可能机制。
文章解读人
王钰玺,上海凡默谷技术部
参考文献
Use of Modeling and Simulation Tools for Understanding the Impact of Formulation on the Absorption of a Low Solubility Compound: Ciprofloxacin.
Marilyn Martinez, Bipin Mistry, Viera Lukacova, Jim Polli, Stephen Hoag, Thomas Dowling, Ravikanth Kona, and Raafat Fahmy. AAPS J. 2016, 4, 886-897. IF: 3.804
参考文献作者单位
美国FDA等
摘要
目的:本研究采用机制性吸收模型探索低溶解度/低渗透性BCS IV类药物在健康受试者的体内行为。
方法:16例健康受试者通过随机交叉的给药设计开展了临床PK试验,试药:一种静脉滴注给药,三种制剂处方口服给药。将环丙沙星口服片在不同溶出条件下的体外溶出试验数据输入到机制性的吸收模型中,探索环丙沙星的体内溶出和肠道不同区域吸收的关系。
模拟结果:尽管环丙沙星的溶出速率会影响药物的释放区域,但该药限制吸收的主要因素是小肠下端及结肠的渗透限速;药物在小肠上端的溶解总量与制剂生物利用度存在明显的相关性;此外,结果表明不同个体的肠道中存在不同的吸收窗口,导致出现吸收窗口差异的原因可能与药物在肠道中发生沉淀有关。虽然,本项目中的模型没包含转运体的影响,但通过几种不同的制剂确定了药物的吸收位置,为研究处方类似的低溶解度/低渗透性化合物时应考虑哪些因素提供了参考。采用机制性吸收模型考察个体的体内PK曲线而不是采用均值曲线,这对研究和解决患者群体服用不同制剂可能遇到的问题很有帮助。
1
项目概述
低溶解度/低渗透性BCS IV类化合物的制剂处方优化面临较大的挑战性。通过机制性吸收模型可以了解制剂处方在体内溶出和体内吸收的情况,深入地理解该药品的体内行为,可能存在的吸收窗口;从而增强我们对制剂处方与患者人体生理学之间相互作用的认知。
本研究目的是探索机制性吸收模型如何指导低溶解度/低渗透性化合物口服制剂的开发。模型的搭建和验证需要以下数据:药物的物理化学特性、基于静脉给药的药代动力学(PK)信息(用于计算清除和分布容积)、口服溶液剂的PK信息(用于表征药物在胃肠道GI溶解后的吸收特性)。通过机制性吸收模型,更好地理解了为获得合适的患者体内疗效,制剂处方设计时应考虑哪些关键因素。
本研究模型药物为低溶解性/低渗透性BCS IV类药物环丙沙星,它具有两相溶解度(在低和高的pH条件下溶解度较大,而中性条件下溶解度较低)。本研究使用GastroPlus™软件构建了正常人体静脉和口服(两种片剂、一种口服溶液剂)模型,在模型中影响药物渗透的因素通过吸收放大因子(ASF)进行考虑。使用计算机模型拟合每个受试者的PK数据,为探究不同处方在各个受试者体内的吸收趋势提供了可能性。
研究环丙沙星体内吸收速率的限制因素,可了解环丙沙星口服制剂设计的关键因素。另外,当制剂用于生理学上不同的患者群体时,由于患者生理差异也会导致药物在体内的暴露量不同,这也是制剂处方开发时需要考虑的问题。
2
建模数据与处理
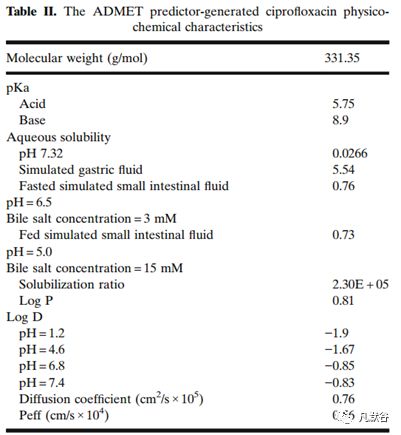
2.1 环丙沙星的相关建模参数
2.2 制剂处方与溶出数据
2.2.1 环丙沙星制剂处方:
1)静脉溶液剂,市售静脉滴注制剂
2)口服溶液剂,市售静脉滴注制剂
3)口服快速释放片剂:粉末直压,处方:羟丙基纤维素(w/w=2)、淀粉(w/w=5)
4)口服慢速释放片剂:粉末直压,处方:羟丙基纤维素(w/w=6)、淀粉(w/w=4)
2.2.2 溶出方法:
USP II,50 rpm,温度为37 ± 0.5°C
2.2.3 溶出介质:
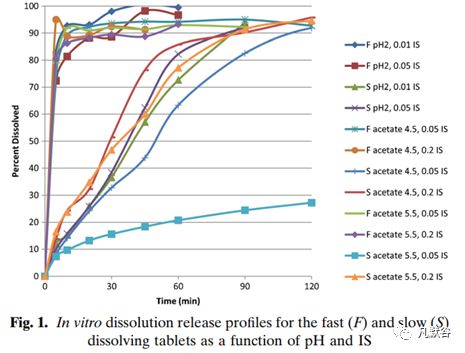
pH 2的HCl溶液(0.01N)、pH 4.5和5.5的醋酸缓冲液,离子强度为0.01、 0.05和0.2,体外溶出结果见下图。
2.3 数据获取及处理
物理化学参数:pKa、溶解度、Log P、分子量、渗透性Peff、胆酸盐效应、扩散系数通过ADMET Predictor™ version 7.2预测得到,溶出模型为Johnson模型。
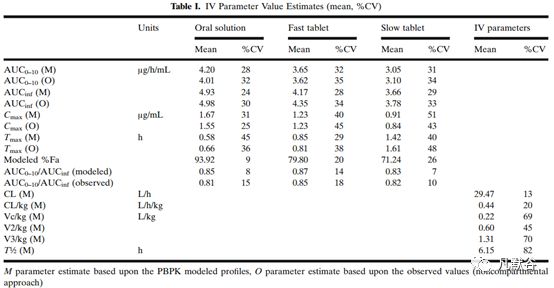
胃肠道模型:使用GastroPlus V9.0默认的高级房室吸收与转运模型,然而环丙沙星的口服吸收受到转运体的影响,为了准确地表征环丙沙星在各个肠段的吸收情况,本研究结合每个受试者的PK数据优化了对应的吸收放大因子(ASF);其中使用口服溶液剂的PK数据优化了十二指肠、空肠的ASF值,使用口服慢速释放制剂的PK数据优化了回肠、盲肠、结肠的ASF。
处置参数:采用2房室或3房室模型,具体的处置参数由每个受试者的静脉给药后的PK数据计算得到。
3
结果与分析
3.1 体外溶出
快速释放制剂的体外溶出受溶出介质的pH和离子强度影响不大,10 min均可释放85%以上,而慢速释放制剂受溶出介质的影响较大。
PK参数拟合结果如下图:
3.2 体内外关系(IVIVR):
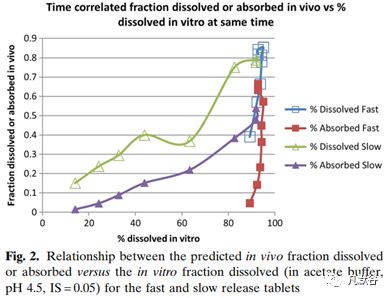
只有慢速释放制剂的pH 4.5醋酸盐缓冲液最能够反映体内PK;而快速释放制剂不具有相关性(体内释放之前体外已经几乎完全释放),如下图,该图反映了制剂的体内溶出和体内吸收的相关性。
3.3 吸收肠段的预测:
药物的吸收主要在前两小时,而这个时间段药物从胃转运到了回肠1段(J1),超出回肠1段药物的吸收非常少,如下图。所有处方都是体内的溶出显著快于吸收,因此是吸收限制了药物的生物利用度。用机制性的吸收模型拟合每个受试者的PK曲线,发现吸收达到最大值时药物的溶出的量在减少,这可能是由于末端肠段药物的吸收较差、且流体体积的减少使得已经溶出的药物在肠道内产生沉淀所致。
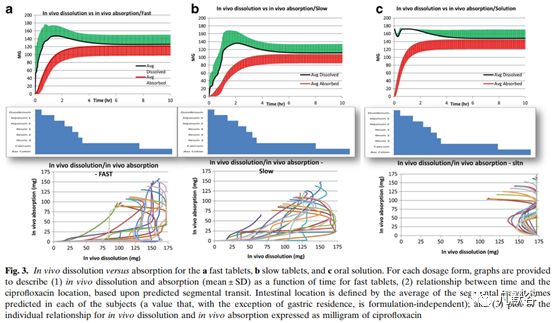
3.4 每个受试者的PK数据分析结果:
并不是所有的受试者都出现了体内溶出逆时针滞后现象(counterclockwise hysteresis),为了进一步研究具有逆时针滞后现象的受试者是否是低环丙沙星生物利用度的受试者,将16名受试者按照环丙沙星的吸收百分数进行了分类,分为以下四类:
吸收较好(所有剂型都吸收70%以上):受试者1、8;
吸收较差(溶液剂吸收小于70%或慢速释放制剂小于50%):受试者2、5、7、10;
吸收不稳定(慢速制剂的吸收大于快速释放制剂的吸收):受试者3、12、17;
吸收均等(或其他):受试者6、9、11、13、14、16、18
分类结果如下图:
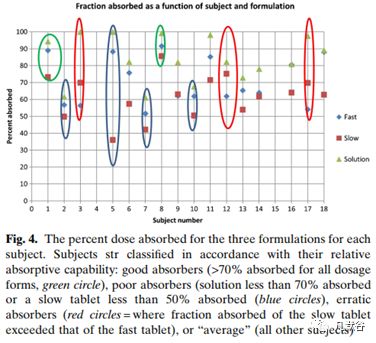
在所有受试者中有6名受试者的药物吸收超过90%,11人吸收大于80%,这与环丙沙星的首过效应较小的结论一致。对于口服3个处方都吸收较好的受试者,没有出现逆时针回滞的情况;吸收较差的一类均有逆时针回滞的情况,然而仅考虑逆时针回滞情况并不能区分吸收均等和吸收较差的受试者,如下图。
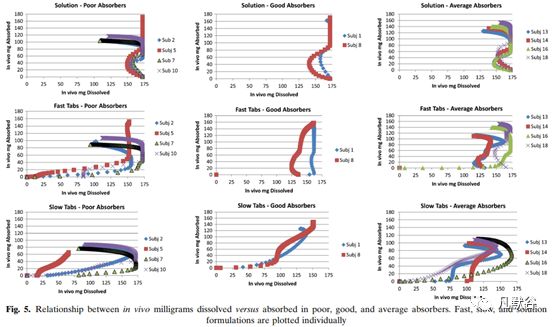
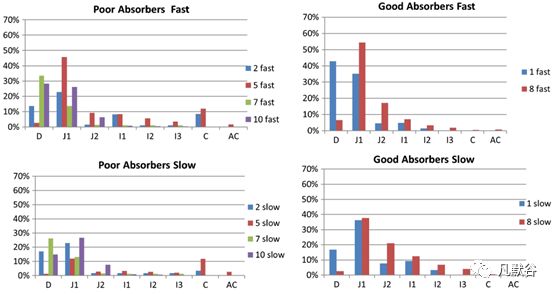
为了更加清晰地理解内在的原因,又考察了每种类型受试者在各个肠段的药物吸收情况,如下图。对于吸收较好的受试者相对吸收较差的受试者在小肠上端有更好的溶出与吸收,吸收较差的受试者主要表现出在小肠上端吸收较差,未吸收的药物转运至回肠和大肠,而在这些肠段环丙沙星的吸收也很差。与吸收较差受试者相比,吸收均等的受试者似乎在空肠肠段2(J2)吸收更多药物,并且在十二指肠(D)和空肠1肠段(J1)中具有相似但略低的吸收,如下图。

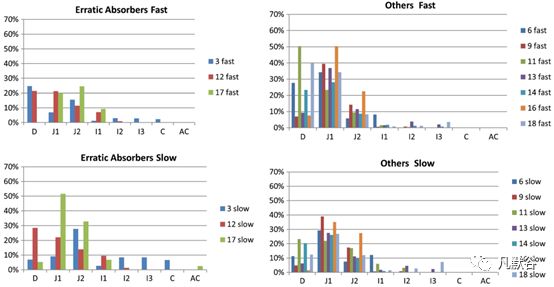
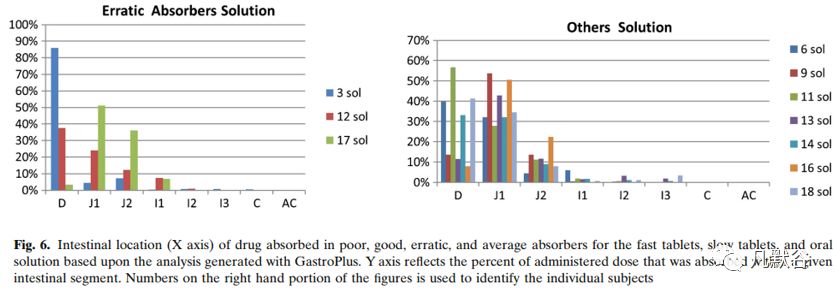
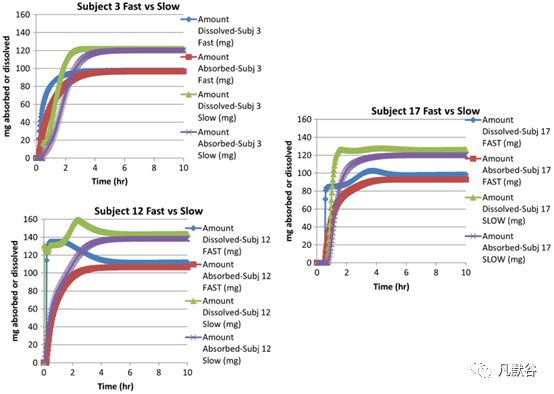
受试者5的情况比较特殊,由于慢速释放制剂的生物利用度较差,将其归到吸收较差的一类,但是该受试者对口服溶液剂几乎达到完全吸收。对于吸收不稳定类(受试者3、12、17),慢速释放制剂的体内溶出超出了快速释放制剂,从而导致更高的药物吸收,慢速和快速释放制剂的体内差异主要出现在前两小时,如下图。因此尽管在很多受试者体内,快速释放制剂溶出的量超过了慢速释放制剂,但是和体内暴露最相关的是前2小时体内药物溶出的总量。
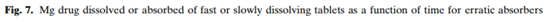
4
模型讨论
机制性吸收模型为探究药物制剂的体内行为提供了可能,借助机制性吸收模型本研究探究了药物制剂在吸收较好、吸收较差、吸收中等受试者体内的溶出的量与溶出部位,考察了吸收和溶出的关系。发现如果环丙沙星制剂不能在小肠上端溶出,则很难具有较好的口服生物利用度。环丙沙星在空肠以下的吸收较差,肠道中肠液体积减少、环丙沙星pH驱动的溶解度下降导致无法将环丙沙星维持在溶解状态,从而使得已溶出的药物减少,出现回滞现象。
第一个回滞现象出现在胃部(10-30分钟),这与已溶解的环丙沙星在到达高pH的十二指肠溶解度下降有关。有报道(Yu et al.)指出环丙沙星的pKa 1在6.0-6.15之间,pKa 2在8.66-8.80之间,在pH 5.5和6.5之间溶解度快速下降,在pH 7.0 - 8.5维持较低的溶解度,这也与ADMET predictor软件的预测结果一致。
第二个逆时针回滞现象出现在空肠以下,本研究认为导致这种现象的原因是肠道肠液体积减少。例如,基于GastroPlus软件中默认胃肠道模型中体积、半径和长度,计算得到十二指肠、空肠1和空肠2的流体体积分别为1.96、1.87和1.65 mL/cm2,对于回肠1、2、3段,流体体积分别为1.46、1.25和1.06 mL/cm2,对于盲肠和升结肠,液体体积分别为0.87和0.77mL / cm 2,这也有文献报道一致(Jakubiah et al.)。第二个逆时针回滞现象的出现也可能是由于吸收的药物外排导致,有报道指出环丙沙星从浆膜到黏膜的转运体含量在盲肠和回肠中有所增加,并且存在明显的个体化差异,这可能也是受试者5慢速释放制剂的低生物利用度的原因。
从制剂处方开发的角度来看,空肠2以下肠段对环丙沙星的吸收将阻碍其缓释制剂的开发。有趣的是,这种解释与以前的报道一致,即环丙沙星吸收的主要部位位于小肠的上部。事实上,目前市售的环丙沙星迟释片剂的平均Tmax为1.5至2小时,这就是为什么胃滞留制剂被探索作为提供环丙沙星持续释放制剂的潜在原因。
对于不同受试者口服溶液剂生物利用度不同的原因可能是由于OATP转运体的密度或活力不同导致的。本研究另一个重要的结果是发现下端肠段的渗透性可以显著影响BCS IV药物的体内行为,虽然本研究受试者的下肠段对环丙沙星吸收较差,但是研究预计,当在临床前物种如狗(回肠和大肠的长度明显短于人类)进行研究时,肠道吸收较低的问题会被放大,在后续的文章中将研究相同制剂在犬体内的溶出和吸收,并与人体机制性吸收模型预测结果进行比较。
尽管增加体内的相关研究将能更加清楚地认识药物体内溶出和吸收的关系,但本研究也取得了一些可靠的结果:(1)仅研究平均数据的会掩盖由于患者本身生理的差异导致药物制剂体内暴露的差异;(2)采用机制性吸收模型的分析方法比仅通过数值分析的方法更能够深入地认识药物制剂的体内行为;(3)下端肠段似乎显著影响某些化合物的吸收;(4)对于可能在肠道内发生沉淀的药物,小肠上部吸收窗口特别容易导致药物吸收的问题,但可以通过调整处方降低此事件发生。
5
结语
本研究使用环丙沙星的物理化学性质数据,静脉、口服溶液剂、口服快速和慢速释放片剂的PK数据,研究环丙沙星口服制剂的体内行为。由于药物摄取和外排转运体和药物代谢途径等变量未纳入该模型,因此当前的机制模型还不能预测不同类型患者(转运体和代谢酶表达不同)体内的药物暴露。
本研究使用高级胃肠道吸收与转运ACAT模型研究药物制剂的体内行为,得到药物在不同肠段的吸收和溶出的可视化特征,从而可确定影响环丙沙星口服生物利用度的关键制剂变量。此外,使用建模和模拟能够考察不同受试者及人群中体内剂型行为的潜在差异。通过分析每个受试者的体内数据,分析不同受试者吸收的差异,可更好地理解药物制剂在各种可能遇到的生理条件的行为,从而指导临床实践。
6
应用软件与模块
本案例应用的软件是GastroPlus (version 9.0),涉及模块有Base, ADMET Predictor, PKPlus, Optimization。
参考文献
英文原文:
Use of Modeling and Simulation Tools for Understanding the Impact of Formulation on the Absorption of a Low Solubility Compound: Ciprofloxacin.
Marilyn Martinez, Bipin Mistry, Viera Lukacova, Jim Polli, Stephen Hoag, Thomas Dowling, Ravikanth Kona, and Raafat Fahmy. AAPS J. 2016, 4, 886-897. IF: 3.804