
新药研发中群体药动学/药效学研究的一般考虑
来源
中国临床药理学与治疗学 2019 Nov; 24( 11)
作者
马广立,许羚,陈锐,陈渊成,赵维,刘东阳,焦正,李健,季双敏,李丽,李良,王玉珠,杨进波,王亚宁,孙鹤,胡蓓,郑青山,卢炜
北京协和医院
复旦大学附属华山医院
北京大学第三医院
NMPA 药品审评中心
上海交通大学附属胸科医院
FDA 药品审评中心
北京大学第三医院
北京大学药学院
江苏恒瑞医药股份有限公司
天士力医药集团股份有限公司
上海中医药大学
山东大学
摘要
群体药动学/药效学研究以及基于此之上的量效关系研究已成为新药研发与监管的工具与重要组成部分。
将其有机地嵌入新药研发和评审过程中,将能够使研发和评审更为高效,并回答一些传统药动学/药效学无法回答的问题。
作为专家共识,本文对群体药动学/药效学研究在新药研发与评审中的应用、试验设计、模型分析方法、分析结果的质量控制与研究报告内容进行了论述。
作者们期望本文能为群体药动学/药效学研究在我国新药研发中的应用提供积极有益的参考。
关键词
新药研发; 定量药理学; 群体药动学; 药效学; 量效关系; 专家共识
1
概述
中国新药研发能力近年来得到了大幅提升,同时定量药理学在新药研发和监管中的重要作用也达成了专家共识[1]。
定量药理学的重要分支———群体药动学/药效学(population pharmacokinetic/pharmacodynamic,PopPK/PD) 及基于此建立的药物暴露-效应(exposure-response,E-R) 关系能够支持制定新药临床研发策略并为科学监管提供关键依据,因此逐步成为新药研发不可或缺的研究内容。
美国食品药品监督管理局(Food andDrug Administration,FDA) 于1999 年首次发表并于2019 年更新了群体药物动力学研究指南[2-3]。
考虑到PopPK/PD 研究的重要性,当前递交FDA有系统暴露的新药注册申请几乎全部包含Pop-PK/PD 内容。
日本药品与医疗器械管理局(Pharmaceuticalsand Medical Devices Agency,PMDA) 于2019 年发表了PopPK/PD 分析指南[4]。
中国国家药品监督管理局(National Medical Products Administration,NMPA) 已经发布药代动力学相关指导原则[5]以及在儿科与抗菌药物相应指导原则中重点提到PopPK/PD 研究的应用[6-7]。
鉴于PopPK/PD 研究在新药研发中的重要作用及当前中文技术文档的缺乏,中国药理学会定量药理学专业委员会组织国内外专家撰写了此文,以期作为我国PopPK/PD 研究的专家共识,进而推动与规范PopPK/PD 研究在新药研发中的应用。
本文从以下五个方面进行了阐述:
①PopPK/PD 研究的基本概念及其在新药研发与监管中的应用原则;
②新药研发中PopPK/PD 研究的一般考虑,包括临床各个阶段数据解读与相关的临床试验设计;
③PopPK/PD 模型分析的一般方法,包括分析计划、模型建立、不同临床应用场景模拟等;
④PopPK/PD 模型分析质量控制与报告的一般考虑;
⑤新药研发中的量效关系研究概述。
1.1 PopPK/PD 研究的基本概念
群体方法(population approach) 与PK/PD 理论的结合形成了PopPK/PD 研究方法[8]。
群体方法的引入使药物的PK 和PD 特征在人群中的变异和引起这些变异的因素得以鉴别与量化[9-10],进而实现了制药行业与监管机构所关注的在人群亚群中对药物的安全性和有效性进行更高效和真实的评估[11]。
群体方法在药物研发中最常用的方法包括两阶段法(two stage approach) 和非线性混合效应模型法(nonlinear mixed effects modeling approach)[2]。
两阶段法先采用非房室分析或房室模型等方法计算每例受试者的PK 或PD 参数,然后通过统计方法(图示法、方差分析或相关性分析等)探索并估算PK/PD 参数与各个潜在因素的相关性[2]。
非线性混合效应模型又可称为分层非线性模型(hierarchical nonlinear model) 。
混合效应泛指固定效应与随机效应,非线性是指这两部分都可以以非线性模型方程式描述。
非线性混合效应模型法是PopPK/PD 研究的核心方法。
相对于两阶段法,基于非线性混合效应模型法的PopPK/PD 研究不仅可以建立PK、PD 学参数的定量关系,同时估计群体典型值、个体间变异与个体内变异,处理和采用稀疏数据,还可以在此基础之上同时鉴别并量化影响PK/PD 参数的协变量因素。
采用非线性混合效应模型的PopPK/PD 模型分析能够定量描述个体的剂量-暴露-效应(doseexposure-response) 关系[12-13]以及协变量对此关系的影响。
协变量通常包括治疗方案因素(如药物剂量、给药间隔、给药路径、合并治疗等)、患者人群的人口学分布情况(如种族、生活地域等)、病理状态(如疾病进程、合并症等)、生理学状态(如体重、性别、排泄和代谢功能等) 等。
以肾功能为例,如果一个药物主要通过肾脏清除,那么同等剂量下药物在不同肾功能的患者中暴露是不同的。
因此,药物在不同肾功能患者中的安全性与有效性也会有所不同。
而肾功能影响不是孤立的,同时又受到患者的性别、体重、种族、是否合并用药等诸多因素影响。
PopPK/PD 研究以肾功能等因素作为分析患者群体的协变量,并进一步量化肾功能等合并因素对剂量-暴露-效应关系的影响。
如果影响因素有临床意义,可以依据PopPK/PD研究结果推荐或调整给药频次及剂量。
基于混合效应模型法的PopPK/PD 研究对于传统PK/PD 不是简单的扩充。
其以下特性更是对传统临床研发方法和理念的变革:
(1) 稀疏: 非线性混合效应模型法使临床试验可以采用稀疏采样的方法获得PK/PD 信息。
较之传统密集采样方法,非线性混合效应模型法使研究临床II 期和III 期更大人群中的药物暴露、有效性、安全性、量效关系、及其变异性成为可能。
(2) 整合: 非线性混合效应模型法可以整合临床前、临床各个阶段的试验信息、药物机理、内部数据或外部文献等信息。
PopPK/PD 研究对试验设计和数据包容性及迭代能力使“学习-确证”(learning and confirming) 等研发理念成为可能。
(3) 分层: 利用协变量解析变异来源,可对群体进行分层,进而实现亚人群的暴露、有效性、安全性的精准量化。
(4) 模拟: 非线性混合效应模型法融合药物、机制、疾病、变异、试验设计等多种因素建立的模型可以进行相对完整的临床试验模拟,进而成为新药研发与评审决策的重要工具。
伴随着非线性混合效应模型的方法理论和软件工具不断提升,以及PopPK/PD 研究的理念逐渐被制药业和监管机构所接受,PopPK/PD 在药物研发中的应用也更加广泛。
1.2 PopPK/PD 研究在新药研发与监管中的应用
传统PK/PD 研究主要关注“整体平均特征”,所以试验设计方面需要尽量避免潜在因素影响。
此类研究通常在受试者特征高度相似的群体中通过密集采样获得该群体的平均PK/PD 特征(例如: 一组18 ~ 40 岁男性志愿者的平均血药浓度-时间曲线和PK 参数) 。
而对于临床研发而言,目标患者人群(如老年患者、女性患者等) 中的变异对临床后续研发以及药物上市后的临床实践极为重要。
评估群体的变异具有重要意义,因为随着变异升高,药物的疗效和安全性在部分人群中可能会下降。
尽早收集并量化群体的信息以及基于PopPK/PD 研究之上的量效关系能够为临床研发策略提供参考,进而从研发途径上提高药物的安全性和有效性。
PopPK/PD 研究在当前临床新药研发和监管中常见的基本应用可举例为以下五个方面:
(1) 剂量选择PopPK/PD 研究及量效关系研究为剂量选择提供关键依据。例如: PopPK/PD 研究得到体重和药物暴露之间的相关性可支持按体重给药或按体重分级给药。
PopPK/PD 研究还可以模拟未经试验过的给药方案下的药物暴露水平。
业内已有成功的案例使用PopPK/PD 模拟未在临床试验中验证过的给药方案,结合暴露-效应关系,证明未验证的给药方案更优并获FDA 批准上市。
(2) 儿童给药剂量与试验设计PopPK/PD 研究对儿童试验尤为重要,因为它可使用稀疏采样尽可能减少采血点,并可使用基于成人数据的PopPK/PD 模型来模拟并支持剂量选择。
对儿童参数的推导应考虑:
①异速放大法;
②生长发育对药物代谢的影响;
③儿童剂型的生物利用度。
应该注意的是: 准确的剂量选择必须建立在对E-R 关系在成人和儿童之间异同的深刻理解之上。
(3) 特殊人群PopPK/PD 研究结果可写进药品说明书,用以支持药物在一般患者人群以及特殊患者人群中的使用。例如: 单独进行肝肾功能不全患者的研究有时可能违背伦理。
这种情况下,可在II、III期的临床试验中纳入一部分肝肾功能不全的患者,并采用PopPK/PD 研究来推荐特殊人群的给药方案。
(4) 药物-药物相互作用PopPK/PD 研究是药物-药物相互作用(drugdruginteraction,DDI) 的重要研究手段。
应用Pop-PK/PD 研究时,建议考虑:
①通常DDI 研究对象是单种药物,而不是一类药物;
②当几种合并用药作用机制和代谢途径相似的情况下,可以考虑将几种药物合并为一个协变量;
③PopPK/PD 模拟可以推荐合并用药所需的最小例数和采样方案;
④对生理上可能相关的协变量需要考虑全面。
(5) 种族差异PopPK/PD 研究可整合多个密集或稀疏采样临床试验的数据,并经过量化得到特定种族人群相关的PK/PD 参数。
基于PopPK/PD 研究进行种族差异评估比传统基于小样本的PK/PD 参数比较更加科学合理。
当不同种族的受试者人群在某些协变量分布上存在明显差异而这些协变量又显著影响PK/PD 参数时,使用PopPK/PD 研究可定量种族与协变量的相关性及影响大小,为阐述种族差异提供证据。
PopPK/PD 研究除了以上五类基本应用,在新药研发中还有更广泛深入的用途。
例如: Pop-PK/PD 研究结合疾病领域知识、临床试验运营等信息能够实现临床试验模拟(clinical trial simulation),为研发决策、试验设计提供证据; 结合疾病机理、转化医学等信息为生物标记物与疾病在临床的相关性提供解释进而促进疾病领域对疾病的认知。
暴露-效应分析作为PopPK/PD 研究的重要应用将在本文的单独章节介绍。
2
新药研发中PopPK/PD 研究的一般考虑
PopPK/PD 研究不仅是一种研究方法和手段,更是一种新药研发的理念。
以下从临床各阶段PopPK/PD 研究的一般考虑以及相关的临床试验设计两个方面予以解读。
2.1 临床各阶段PopPK/PD 研究的一般考虑
按照经典的划分法,临床试验阶段可划分I、II、III、和IV 期。
I、II 期临床试验一般认为是临床早期探索阶段; III 期临床试验为后期确证性的注册试验; IV 期临床试验则为上市后研究。
PopPK/PD 研究在临床不同阶段的考虑也会有所不同。
需要特别注意的是PopPK/PD 研究一般不是基于一个临床试验的独立研究,而是整合当前试验与之前关键PK/PD 数据与信息的综合研究。
新药的早期临床研发需要尝试回答以下两个问题:
①新药在目标人群的剂量-暴露-效应(安全性与有效性)关系;
②内外因素对剂量-暴露-效应(安全性与有效性) 关系的影响。涉及这两个问题的临床试验(首次人体试验、剂量探索等),在临床资源及操作允许的情况下,研究者可以针对研究药物的PK、PD 特性收集相应的协变量信息。
基于收集的信息进一步定量剂量-暴露-效应关系,并探索其可能的影响因素。
新药早期临床研发是一个逐步递进的过程,推荐在首次人体试验的起始阶段就开展PopPK/PD 研究,而后逐步建立剂量-暴露-效应的关系并量化其影响因素。
早期临床研发中的首次人体、特定人群、首次患者等试验可以带来不同信息以支持下一步临床研发策略的制定与更新。
同时,大部分早期临床试验为密集采样,对于理解药物PK 属性具有不可替代的作用。
需要注意的是,由于早期临床研发通常样本量较小,因此PopPK/PD 研究的结论可能有一定局限性。
在新药临床研发后期,关于药物的非临床与临床信息已经基本齐备,再结合III 期临床试验大规模患者群的PK、安全性、有效性数据,PopPK/PD 研究能够为药物研发者和监管者提供相对全面的包含人群亚群的安全性和有效性的确证信息。
当临床不良事件发生时,用PopPK/PD 方法判断其与药物剂量或血药浓度、生理病例特征是否相关,也将成为PopPK/PD 研究应用的重要场景。
基于PopPK/PD 模型能够模拟推演未纳入研究的给药剂量、给药频率等情况下的安全性与有效性信息以支持药品注册和上市后的临床应用。
PopPK/PD 研究对于新药评价、药物说明书、临床指导用药等均能够提供不可或缺的信息与证据。
临床阶段需要强调的是,PopPK/PD 研究成功应用的首要前提是数据质量。
这需要临床团队紧密合作,对给药时间、采样时间等各种涉及到PopPK/PD 研究的变量准确记录,并严格按试验手册规定要求处置相关样本。
只有高质量的数据才可能支持高质量的PopPK/PD 研究。
2.2 PopPK/PD 研究相关的临床试验设计
制药行业与监管机构越来越重视暴露量与安全性/有效性的关系,通常情况下只要试验条件允许都会采集PK、PD 数据进行PopPK/PD 研究。
Pop-PK/PD 研究相关的临床试验设计根据药物所处的不同临床阶段会有不同考虑。
I 期临床试验为了精确定量药物的PK 属性采用密集采样获取血药浓度数据,某些PD 数据(如血糖) 也尽可能进行密集采样。
这个阶段对于理解药物的PK/PD 行为至关重要,也是后续PopPK/PD 研究的出发点。
II 期和III 期临床试验,在大规模患者群中密集采样会变得不切实际。PopPK/PD 研究能够有效利用稀疏采样数据。
设计PopPK/PD 研究相关的PK、PD 指标采样时,需要考虑现实试验情况的局限性,比如采样次数、采样时间、每个受试者的标本数量、受试者例数等。
如果能够根据I 期的密采数据,通过建立的Pop-PK/PD 模型模拟试验设计会使试验设计更为有效。
当受试者例数、每个受试者的标本数量方面存在很大的局限性时(例如儿童或老年人群等),优化采样设计就变得格外重要[14]。
优化采样设计能够使临床试验为PopPK/PD 研究提供更丰富的信息[15]。
以下主要介绍PopPK 相关的试验设计方面考虑,PD 方面因疾病的不同、生物标记物不同、临床指标不同,情况较为复杂,限于篇幅不详细解读,但读者可以此做参考。
2.2.1研究人群
研究者应当依据研究目的选择研究人群,并尽可能包括药物的未来临床拟应用人群。实际操作中,需要试验方案具有明确的入排标准,并在执行层面严格遵守以保证受试者人群的协变量分布能够支持PopPK 研究的协变量分析从而实现研究目的。
亚人群如儿童、孕妇、老年人等变异较大的人群,需要根据人群的特征变量如年龄、孕周期等进行分层入组受试者。
同理,肝功能、肾功能不全等患者,也需要按照疾病严重程度分层入组,以准确全面地评价各个疾病状态的PK/PD 特征。
2.2.2 样本量
由于PopPK 研究可以整合各种信息,并且方法和模型不固定,所以对样本量计算方法没有严格要求。
鉴于PopPK 的研究目的除了准确估计PK 参数及其变异外,还包括定量分析影响因素,因此PopPK 试验应在潜在重要影响因素不同水平纳入受试者进行研究。
研究者通常在临床试验之前可能无法获得全部重要影响因素信息,所以建议临床研究人员尽可能多地纳入受试者并使其多样化,以便能支持临床复杂情况下的剂量优化[15]。
对于重要的待评价影响因素,各个水平的样本量应具有可信的代表性,原则上大于整体研究人群的5%。
2.2.3 PK/PD 指标设计
PopPK 研究目标主要为原形药物,但通常在代谢物有活性且影响药物的剂量-效应关系时也研究其代谢产物。
PopPK研究指标通常指体循环暴露参数,部分研究也可能涉及其他样本(如尿液、唾液、脑脊液等) 中的药物暴露参数,或是通过影像学方法量化组织中的药物浓度所得到的暴露参数。
PD 研究需要考虑选择与药物机理机制相关,并且纳入可客观、定量评价的指标进行分析。
同时,潜在可能影响暴露-效应关系的病理、生理指标也可以纳入研究。
某些PD 指标收集时间点可与PK 类似,特别是需要考虑PD 指标随时间变化的过程时(如血糖、QT、血压等) 。
由于这类PD可能呈现非线性特征,并且可能伴有耐受、反跳、滞后、日间节律、进食影响等现象,所以明晰研究药物的作用机制、靶点的病理生理特征以及疾病进程对PD 的精准定量具有重要意义。
另外一些临床PD 指标是一次性(如感染被治愈)、缓慢渐进的(如血脂改变、记忆力改变)、突发的(如肌腱断裂),PD 指标收集时间点可与一定时间周期内的某些对应时间点的稀疏PK 采样点类似(如谷浓度)。
2.2.4 采样点
临床II 期和III 期研发中应用普遍的两种采样设计:
①谷浓度采样设计(troughsampling design) ;
②全PopPK 采样设计(full populationPK sampling design) 。
谷浓度采样设计是指在药物谷浓度时或接近药物谷浓度时(即下次给药前) 从每个受试者采集血样[16]。
谷浓度尽管包含了一定的吸收和分布信息,但信息主体主要集中在消除相。
由于单个谷浓度采样设计无法评估个体内变异(withinsubject variability),所以在条件允许的情况下应当使用多个谷浓度(推荐至少3 个谷浓度) 采样以保证个体内变异和个体间变异(between subjectsvariability) 能够得到评估。
全PopPK 采样设计有时也被称为实验性PopPK 设计(experimental pharmacokinetic design)。
全PopPK 设计在给药后对每一个受试者多个不同时间点(如最佳采样点optimal samplingtime) 进行采样,通过全部受试者的血药浓度信息来描述PopPK 特征[17]。
对于新药研发而言,推荐采用全PopPK 采样设计以研究目标人群中药物的药动学与人口统计学、病理生理属性之间的关系。
场景间变异(inter-occasion variability) 会影响到协变量、个体内变异、个体间变异的准确估算[18]。
试验设计方面可以通过每一个受试者在多个场景采样来保证至少有一个适度规模的受试者子集提供不止一次的数据以估算场景间变异。
2.2.5 协变量
PopPK/PD 研究开始前,需要详细记录研究人群的人口统计学特征。
同时,依据研究目的收集相关协变量,协变量通常是那些被认为可能影响PK 或PD 的已知因素,如合并用药、基因型、实验室测定指标等。
待研究的协变量选择应该避免盲目广泛筛选,而是基于临床实际情况、药物作用机制、临床药理学意义等进行选择。
协变量一般采用服药前的基线值,但如果协变量在整个研究过程中存在较大改变(如10%以上),则建议在不同时间点采集变化值。
如果协变量变化趋势明显,研究者应该充分收集指标变化的原因,并分析是否是药物作用导致。
若判定是药物引起的指标变化,即使研究初始时将其作为协变量,也应当尝试将该指标作为药物作用的指标进行分析。
3
PopPK/PD 模型分析的一般考虑
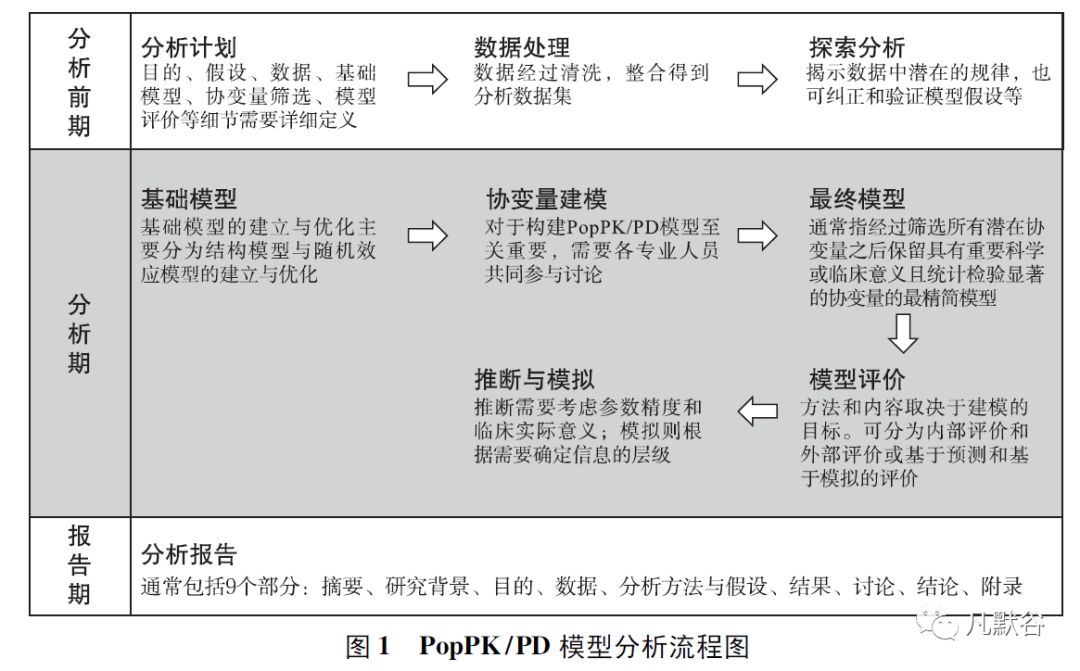
PopPK/PD 模型分析过程可以分为分析前期、分析期、报告期。
如图1 所示,分析前期包含分析计划、数据处理、探索分析; 分析期包含基础模型、协变量建模、最终模型、模型评价、推断与模拟; 报告期的PopPK/PD 模型分析报告为最终交付文档。
PopPK/PD 模型分析软件数量不断增加,其基本理论依据和方法与NONMEM 相似。
虽然鼓励在新药研发过程中应用其它软件进行PopPK/PD 模型分析,但是考虑到NONMEM 一直是行业标准,本文以NONMEM[19]为例进行讨论。
下文就PopPK/PD 模型分析各个环节予以介绍。
3.1 分析计划
PopPK/PD 研究需要多专业进行协作研究以解决临床研发具体问题,因此需要定量药理学工作者和临床研发团队进行沟通,以确定最佳研究方案。
研究者需要提早制定并及时更新PopPK/PD 模型分析计划书。
该计划书应包括研究目标、方法、假设、所需数据和时间表等重要因素,应明确研究数据集、研究方法及结果评价方法与标准,具体要求可以参考定量药理学研究专家共识中的定量药理学分析计划书[1]。
3.2 数据处理
数据质量是PopPK/PD 模型分析的根本前提。
任何数据相关问题和操作都必须严格记录。
为了保证数据处理过程的全程可追踪,推荐使用程序脚本进行数据操作,避免手动处理数据。
3.2.1 数据组成
一般情况下开展PopPK/PD模型分析时,数据集由给药方案、体内暴露量、药效、协变量等数据组成。
给药方案通常包括: 给药剂量、时间、途径等。
体内暴露量通常包括给药后某时间点的血或组织中药物浓度,即PK 数据。
时间数据包括给药、PK/PD 采样等所有事件发生的时间信息。
协变量数据通常包括:
①人口统计学: 性别、年龄、体重、民族等;
②实验室检查数据:如血常规、血生化(肝功能指标、肾功能指标、血糖)、电解质等;
③合并用药情况;
④既往病史;
⑤生命体征: 如月经周期、持续性失眠状态等;
⑥其他: 如药物基因组学数据、家族病史等。
PD 数据通常是分析相应的临床终点指标、可反映研究药物作用机制或与临床终点相关联的生物标记物等。
如果研究目的是分析暴露与不良反应的关系,PD 指标应是分析所关注的不良反应指标。
3.2.2 缺失数据
PopPK/PD 模型分析实践中,缺失数据问题很难避免。
在所有可能找回数据的办法失败后的处理方法:
①时间数据: 采用的临床试验方案时间,或更严格情况下缺失时间信息的PK/PD 数据不纳入分析;
②PK 与PD 数据缺失:一般不予填补。
需要声明的是,使用PopPK 技术外推或模拟得到的PK 数据进行PopPK/PD 分析也是PopPK/PD 分析的方法之一,与这里提到的“数据缺失”不是同一概念。
3.2.3 错误数据
PopPK/PD 模型分析原则上讲不应该剔除任何数据点。
但临床实践中,如果有确凿证据证明数据点采集或生物分析等操作过程存在错误,这类数据归为错误数据。
引入错误数据必然会影响PopPK/PD 模型分析的正确性与科学性。
因此潜在错误数据必须严格排查、记录并归档。
错误数据必须从分析中剔除但仍建议根据最终模型进行敏感性分析。
3.2.4 低于定量下限浓度
一般情况下,低于定量下限(below limit of quantification,BLQ) 浓度数据如果占总数据比例低于10%,可将BLQ 数据忽略不纳入分析。
如果比例高于10%,需要根据具体情况选择恰当的处理方法[20]。
3.2.5 异常值
异常值(outlier,又可称为离群值) 通常情况下以基础模型或最终模型的加权残差(weighted residuals versus time,WRES),条件加权残差(conditional weighted residuals versus time,CWRES) 等大于预设值(如6) 为判断标准。
如果异常值干扰基础模型建立和协变量的筛选,则早期分析中可不纳入异常值。
建议利用最终模型对异常值做敏感性分析。
分析报告中需要总结包含与不包含异常值的结果。
如果异常值对模型参数影响不显著,建议以包含异常值的参数值为最终报告值。
3.2.6 临床试验数据整合
PopPK/PD 模型分析经常需要整合多个临床试验数据。
PK、PD、协变量都可能由于所处研发阶段、试验设计、执行时间、检测方法、涉及人群、所在地域等情况而变化。
整合多个临床试验数据时,需要统一变量单位并且引入一个或多个协变量标注区分不同的数据源以便探索分析和建模分析时予以适当考察或处理。
3.3 探索分析
在开展PopPK/PD 模型分析之前,首先应对数据进行探索分析。
探索分析是一种采用图表化和统计学分析技术探索和揭示数据所蕴含特征的分析方法。
同时,探索分析可以验证模型假设正确与否,以及当假设有误时可指导纠正模型假设[21-22]。
探索分析方法和结果应简要地加入PopPK/PD 模型分析报告。
3.3.1 样本来源图表
分析报告中应简略介绍分析中所包含的每个临床试验,并采用表格和作图的方式总结每个临床试验特征,如试验编号、受试者数目、受试者在试验过程脱落情况、受试药物的种类、剂型和剂量、生物样品的种类和数目、生物样品采样时间、生物样品的分析方法及其检测定量上下限等。
3.3.2 协变量图表
分析报告中应采用表格和作图的方式总结PopPK/PD 模型分析中涉及的协变量。
协变量包括连续型协变量和分类型协变量。
连续型协变量需提供描述性统计结果,并采用直方图描述连续型协变量的总体分布等。
分类型协变量需提供描述性统计结果,并计算每个协变量各分类的频数和频率。协变量也可以在必要时按照亚群体进行分层描述性统计分析。
同时,分析报告中也应该采用图表的形式探索各个协变量之间的相关关系。
3.3.3 因变量作图
PopPK/PD 模型分析中因变量(dependent variable,DV) 通常是指血药浓度、有效性与安全性等药效学指标。
生物样品中的药物及其主要代谢产物浓度、药物在受试者中的有效性与安全性等药效学指标随时间作图以及血药浓度对药效指标作图,对药物的PopPK/PD 特征的理解是非常重要的。
这里用血药浓度为例说明因变量作图,药效学指标虽然更复杂但可以此作为参考。
血药浓度随时间的变化曲线应采用常规坐标和半对数坐标两种形式作图分析。
每个受试者的血药浓度数据点之间可采用连线的方式描述血药浓度随时间的变化趋势。
血药浓度随时间的变化曲线可在必要时按照亚群体进行分层作图分析。
如果对原始血药浓度数据进行任何形式的转换,必须在分析报告中详细阐明并提供合理的论证。
用于内部建模和外部验证的原始数据需分别作图描述。
3.4 基础模型
PopPK/PD 模型分析中的基础模型是指包含结构模型和随机效应模型,且未经协变量筛选的模型。
基础模型的建立与优化主要分为两步: 结构模型的建立与优化、随机效应模型的建立与优化。
3.4.1 结构模型
PopPK/PD 模型建立的第一步需要关注的是基础模型的结构形式,如一室模型、二室模型、药效学中的米氏方程模型等。
模型建立者可使用前期获得或文献报道的PK/PD 信息来帮助进行模型的构建。
基础模型的构建和选择,需要考虑建模目的和模型用途以及数据是否能够支持模型。
若存在已知的、对固定效应参数有特定影响的协变量,建议将其纳入基础模型。例如: 已知对药物代谢有影响的基因型,可考虑作为结构模型协变量。
对药物吸收存在较大影响的因素,如食物或制剂形式,在模型中若不加以考虑,可能会导致模型不稳定。
建模的数据中体重范围较宽或同时包含成人和儿童数据时,建议考虑将异速生长模型作为PK 结构模型的一部分。
3.4.2 随机效应模型
PopPK/PD 模型中,随机效应可划分为个体内变异、个体间变异、场景间变异。
(1) 个体内变异个体内变异,又称为残差(residual unexplainedvariability,RUV) 变异,采用ε 表示,一般假设其服从均值为0,方差为σ2 的正态分布。
残差模型的选择需要考虑PK/PD 数据的特征。
这些特征包括PK 数据是否作对数转换,PD 是否是连续、分类、或时间事件类型数据等。
残差模型会严重影响模型结构、个体间变异等评估。
通常情况下,应当在建模初始阶段根据PK/PD 数据特征决定残差模型结构,比如PK 残差模型采用加和、比例、或是加和比例结合模型。
(2) 个体间变异个体间变异,一般采用Ω 表示,并假设其服从均值为0,方差为ω2 的正态分布。
PopPK/PD参数一般情况下不会出现负值,所以建议采用指数函数形式描述IIV。
IIV 的估算需要考虑数据是否能够支持。
以一室口服吸收的PopPK 模型为例,通常在CL 和V 上引入IIV。
但若吸收相采集了丰富的数据,也可在描述吸收的参数如Ka上尝试引入IIV。
IIV 参数计算值接近0 时,并不意味着该参数不存在个体间变异,而是表明数据可能不足以估算IIV。
在探索IIV 时,个体间随机效应的相关性也需要谨慎评估。
当个体间随机效应存在相关性时,可能会导致参数估算不准确。
(3) 场景间变异场景间变异(inter-occasion variability, IOV)可能由试验设计如交叉试验设计、试验周期等因素产生。
例如: 在I 期交叉、剂量递增试验中,场景通常由周期定义。
在II、III 期试验中随访时间较长,场景可以定义为PK 参数可能改变的时间范围如周、月、年等。
整合来自不同试验时期数据进行建模时,应谨慎考虑来自不同阶段数据对于场景的定义不同,可能会导致IOV 的解释变得困难。
IOV 可在协变量筛选前即加入基础模型中,但建议在整个建模过程中评估IOV。
PopPK 基础模型应足以描述研究药物的典型药时曲线; 其统计模型可描述典型药时曲线的变异特征(个体内变异、个体间变异、和场景间变异) ; 同时建议纳入已知的协变量,如采用异速生长模型将体重固定于CL 和V。在基础模型构建的过程中,需要始终以研究目的和用途为导向。
3.5 协变量
建模协变量建模需要谨慎面对过参数化、协变量间的相关性/共线性以及不均衡的协变量。
如果能够在引入协变量之前,从以下几个角度审查协变量会对协变量分析起到重要作用:
①科学性: 利用生物学、药理学、病理学、医学等专业知识审核协变量和模型参数的潜在关系;
②先验知识: 临床前信息、文献信息、早期临床证据等;
③图形探索分析: 例如血药浓度、非房室分析参数(如曲线下面积或最大血药浓度等) 与协变量的关系。
缺失协变量值是实际分析中经常遇到的问题。
在无法找回缺失值的情况下,一般采取以下方法进行处理:
(1) 所有缺失协变量值的受试者数据不纳入分析。
(2) 使用推算值替换缺失协变量:
①协变量的中位数替换缺失协变量值;
②对于同一个受试者,使用基线、前一时间点或其他时间点值替换缺失协变量值;
③使用插值法推算协变量值。
(3) 如果大比例缺失协变量值,协变量不适合纳入分析。
PopPK/PD 模型分析中的协变量建模在保证能够描述数据趋中性的同时,应尽可能保持简洁。
通常情况下,推荐采用连续变量的中位数或分类变量的最普遍分类作为参考值。
协变量可以以加和、比例、或是指数加入模型。
协变量的统计分布需要在加入模型之前就予以评价。
协变量加入模型时建议以特定值做中心化。
中心化协变量可能使参数解释更容易、并提升参数估算时的数值稳定性。
协变量模型要考虑数学、统计、科学上的可能性并避免出现现实世界不可能发生的参数值或假设。
纳入协变量的方法主要有两种:
①筛选协变量法: 利用中间步骤,即从基础模型开始逐步筛选、评价、加入或删除协变量直到形成最终模型;
②全协变量法: 将所有可能相关的协变量同时包含到模型中,从而生成最终模型。
筛选协变量法包含多种方法。
逐步协变量建模(stepwise covariate modeling,SCM) [23]由于概念简单、操作方便、自动化工具完备,是筛选协变量法中应用最普遍的方法。
SCM 首先前向选择协变量加入模型然后后向删除协变量形成最终模型。
筛选协变量方法还包括沃德约化建模(Wald's approximation modeling,WAM) [24]、广义加和模型(generalized additive model,GAM) [25]、Lasso 建模(least absolute shrinkage and selectionoperator, lasso) [26]等。
全协变量法(full model estimation,FME) [27]指同时加入全部潜在协变量,一步完成协变量建模达到最终模型。
由于没有过多的建模步骤,能够使协变量评价过程更加可控。
相对于变量筛选法,此方法能够直接评估并显示所有潜在协变量作用的大小。
全随机效应法(full random effectsmodel,FREM) [28]有效解决了FME 关于协变量间的相关性问题,正逐渐被业内所接受。
协变量建模在有些情况下是可以省略的,并以基础模型为最终模型。
这些情况包括但不限于受试者人数较少、人群均一、试验设计不适合考察协变量等。
在协变量不具备预测价值的情况下,这种不包含协变量的最终模型甚至可能会比包含协变量的模型具有更好的预测性。
3.6 最终模型
最终模型通常是指经过筛选所有潜在协变量之后,保留具有重要科学或临床意义且统计检验显著的协变量的最精简模型[29]。
随着学科发展,最终模型的概念正在发生变化。
比如,比例缩放方式加入结构模型的体重未必具有统计学意义; 全随机效应法模型的协变量未必最精简。
理论上讲,最终PopPK/PD 模型必须能够充分描述化合物的PK/PD 属性及变异性的来源,并且具有一定的预测能力。
3.7 模型评价
模型评价的方法和内容取决于建模的目标。
如果建模目标只是描述数据特征,则使用的模型评价方法可相对较少; 若目标为预测,则通常需要更为全面和严格的模型评价。
根据用于评价的数据来源,模型评价可分为内部评价(internal evaluation) 和外部评价(external evaluation)。
按照具体实施手段,模型评价又可分为基于预测(prediction-based) 和基于模拟(simulationbased)的评价。
3.7.1 内部评价和外部评价
内部评价指评价的数据集与建模的数据集来自于同一研究。
常用方法包括数据分割法(data splitting) 和重抽样法(resampling techniques)。
后者在群体研究中应用更为广泛。
数据分割法适用于大样本量的数据集。
通常在建模之前随机选择部分数据作为建模数据集,此部分数据通常为原始数据集的70% ~ 80%,然后将剩余的20% ~ 30% 数据作为评价数据集,用于评价所建立的模型。
制定分析计划时,应明确定义数据分割的方法,确保评价数据集具有代表性。
数据分割时,以单个研究对象为最小分割单位,一般不将单个研究对象的数据分成两部分。
此外,数据分割时应考虑研究的试验设计、采样策略与研究对象的特征,必要时应作分层,以保证建模数据和评价数据集中不同类型特征的数据具有相似的比例。
数据分割法中,建模数据样本量的减少会降低构建模型的预测精度。
交叉评价法(cross-validation)[30-32]和自举法(bootstrap) [33]等重抽样技术能够充分利用数据。
交叉评价法可视为重复多次的数据分割,与数据分割相比,其优势在于充分利用了所有数据; 其缺点在于估算准确性存在较大变异,且重复多次的评价过程往往较为低效[2]。
自举法是一种有放回的重抽样方法,是目前应用最为广泛的PopPK/PD 模型分析的评价方法之一。
自举法能较好地评价模型参数估算值的可信度和模型稳定性,但不能反映模型对研究数据集的拟合优劣程度,以及模型的预测性能。
外部评价指应用独立于建模数据之外的数据集对模型进行评价。
如果建模数据来源于多中心的大样本数据,可仅采用内部评价。
当建模数据来源于单中心研究时,除内部评价外,还应进行更为严格的外部评价。
内部评价和外部评价的本质区别在于评价数据集的来源。
鉴于不同的研究之间可能存在一些无法控制的差异,在解读外部评价结果时应重视这些差异,不理想的评价结果并不意味模型不当。
如果基于外部评价的结果是理想的,则外推预测成功的可能性也将增加。
与内部评价一样,选择评价数据集时,应考虑研究设计和研究对象的特征是否与建模数据集存在差异。
3.7.2 基于预测和基于模拟的评价
基于预测的模型评价是指将绘制模型诊断图、计算预测误差(prediction error,PE) 相结合的方法,比较模型预测值与观测值,从而综合评价模型预测性能。
模型参数的估算过程中,可采用不同的算法,如一阶估算法(first-order estimation,FO)、一阶条件估算法(first-order conditional estimation,FOCE) 等。
此外,预测值包括个体预测值和群体预测值。
因此,在进行比较和评价时,应对采用的算法和预测值的类型加以说明,解读结果时,也应作相应的具体分析。
基于模拟的模型评价是指通过建立的模型及参数,进行蒙特卡洛模拟,生成若干套模拟数据集。
通过诊断图结合统计学检验的方法,综合评估模拟数据与观测数据分布特征的相符程度。
如果模型对原始数据的拟合程度较高,较为准确地描述了原始数据的分布特征,则基于模型的模拟数据应能较好地再现原始数据的分布;
反之,如果基于模型的模拟数据分布与原始数据存在较大偏差,则提示模型需要进一步优化和改进。
3.7.3 常用评价方法
模型评价方法的选择取决于建模目的,评价结果的呈现往往采用诊断图和统计学检验相结合的方法。
常用的诊断图包括拟合优度诊断图、预测误差检验、可视化预测检验(visual predictive check,VPC)、数值预测检验(numericalpredictive check,NPC) 等。
不同的评价方法通常仅能展现模型在某一方面的特征,因此在实际应用中,常常采用多种评价方法相结合来对目标模型进行综合评估。
3.7.3.1 诊断图
诊断图是最为常见的一种模型评价方法,如对基础模型和最终模型的比较与评价。
模型诊断图包括: 基于预测的模型诊断图、基于残差的模型诊断图、基于经典贝叶斯估算的模型诊断图[29]。
不同的诊断图从不同的视角评估模型的准确性与适用性,反映了模型化过程中的模型设定错误、违背随机变量分布假设以及离群值等问题。
这些诊断图可并行比较,描述纳入协变量前后模型的改善情况,以指导模型的开发与优化过程。
基于预测的模型诊断图侧重呈现实际观测值与群体/个体预测值的一致性,直观地反映模型对观测数据的拟合程度。
基于预测的模型诊断图主要包括:
①因变量-群体预测值(dependent variableversus population prediction,DV vs.PRED) 图,可直观地评估群体预测值能否很好地描述数据的集中趋势和离散程度。
如图2(本章节图例源自参考文献[34]) 所示,x 轴为基于模型的群体预测值,y 轴为因变量/观测值,虚线代表参考线,实线代表趋势线。
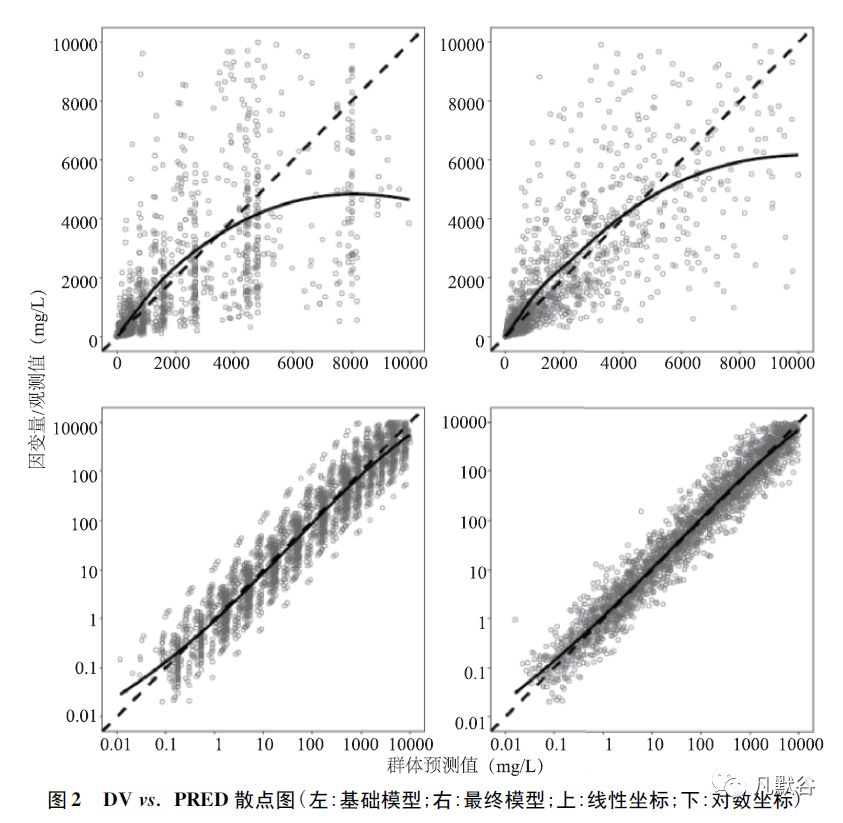
与基础模型(左上、左下) 相比,最终模型(右上、右下) 的拟合优度大大提高,最终模型较好地描述了数据的集中趋势;
②因变量-个体预测值(dependent variable versus individual prediction,DV vs.IPRED) 图,可直观地评估模型的个体预测值能否很好地描述个体观测值的集中趋势和离散程度,各数据点在对角线的分布情况反映了残差变异的分布和大小。
如图3 所示,x 轴为基于模型的个体预测值,y 轴为因变量/观测值,虚线代表参考线,实线代表趋势线。
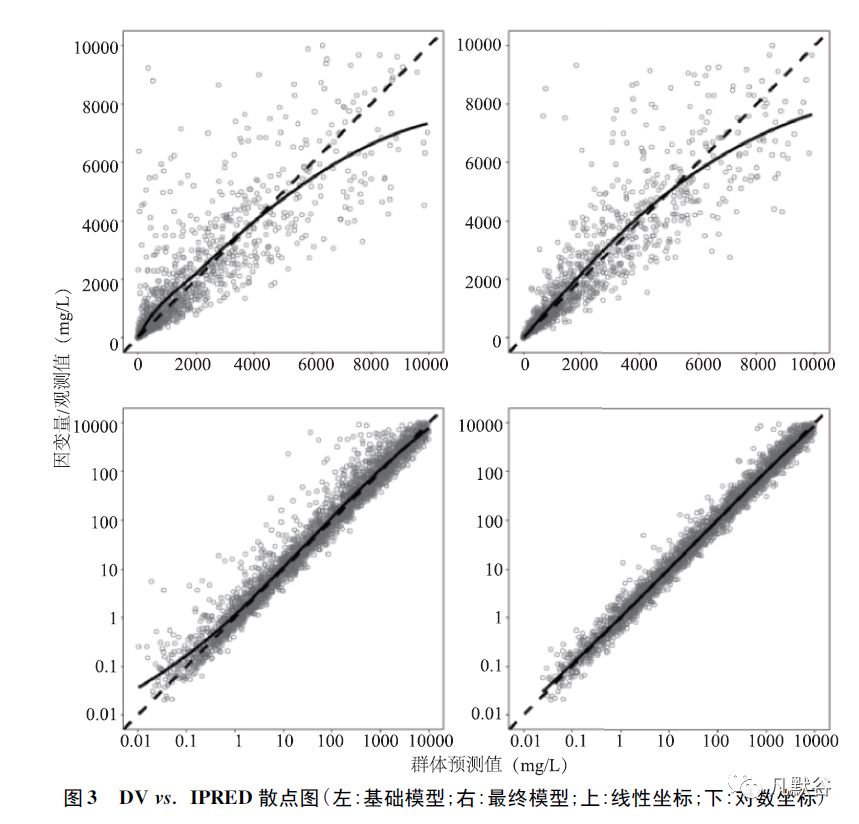
基础模型(左上、左下) 与最终模型(右上、右下) 的DVIPRED 诊断图基本相近;
③个体药-时曲线图,可直观地评估模型对于每个个体的拟合程度。
如图4 所示,x 轴(自变量) 为末次给药后时间,空心圆点代表每个个体的实际观测值,虚线代表基于模型的群体预测值,实线代表基于模型的个体预测值。
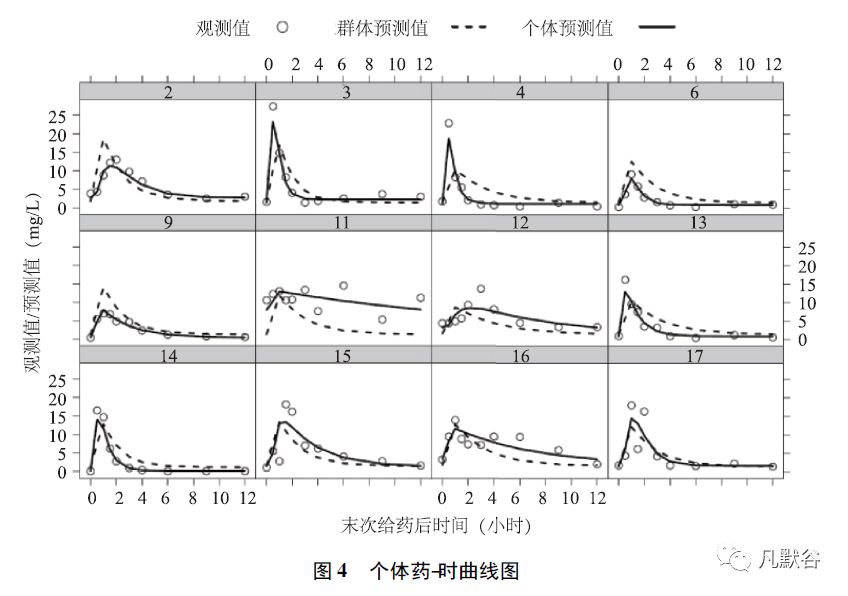
个体预测值的拟合程度明显优于群体预测值,对多数个体而言,模型对峰浓度的拟合情况不佳,提示可能需要进一步优化改进吸收模型。
相对于基于预测的模型诊断图,基于残差的模型诊断图可以更为直观地评估这些预测偏差。
不同的残差诊断图往往能够显示不同结构模型或统计学模型的特性。
为了更充分地评估模型的准确性与适用性,建议除了一致性诊断图外,应更加重视残差诊断图在模型评价中的作用。
基于残差的模型诊断图主要包括:
①WRES 图,对自变量诊断图可用来评估结构模型的准确性;
②条件加权残差-预测值(conditional weighted residuals versusprediction,CWRES vs.PRED) 图,一定程度上要优于WRES vs.PRED。
如图5 所示,x 轴为基于模型的群体预测值,空心圆点代表条件加权残差,虚线代表参考线,实线代表趋势线。
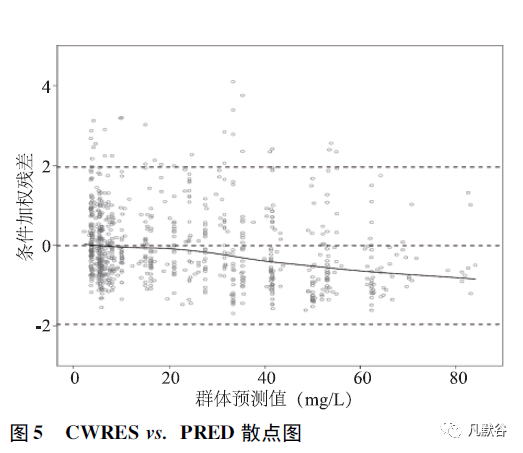
绝大多数的条件加权残差均分布在± 2 之间,随群体预测值增加,条件加权残差呈现明显趋势性变化,提示模型对高浓度数据拟合欠佳;
③条件加权残差直方图和Q-Q(quantile-quantile plot) 图,可识别残差是否服从以零为中心的单峰对称分布。此外,亦可用Q-Q 图来描述残差的分布特征。
如图6 所示,直方图中虚线为核密度曲线,Q-Q 图中实线为y= x 参考线。
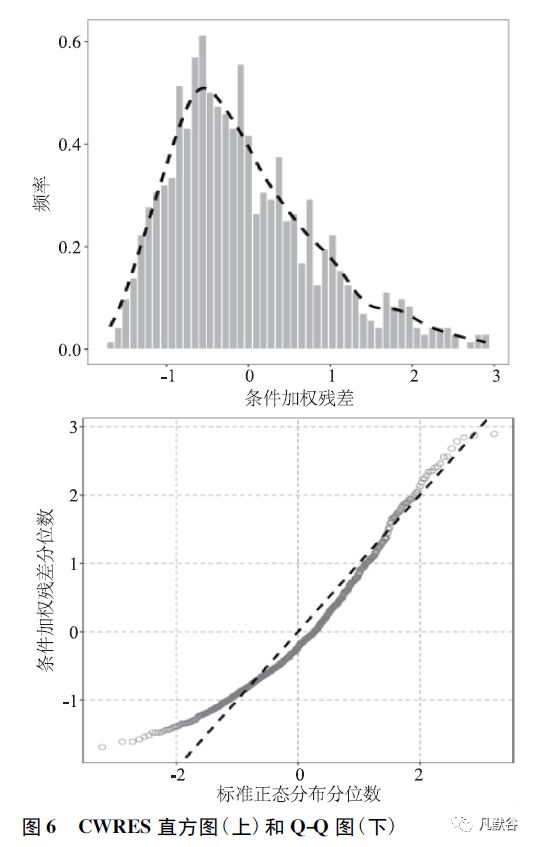
若条件加权残差服从正态分布,则空心圆点应近似分布在参考线附近。
由图可见,空心圆点在高群体预测值时偏离参考线,提示条件加权残差不完全服从正态分布。
应用诊断图对模型进行评价时,须警惕“观测值与个体预测值完美吻合”的假象。
如果数据能提供充分的信息,参数水平的个体间变异和观测水平的残差变异均服从标准正态分布。
当数据稀疏且个体观测信息不足时,个体参数的经验贝叶斯估算值将趋近于群体均值,此时个体间变异的分布将趋近于0,称之为个体间变异收缩(η-shrinkage) 。
类似地,随着数据量的减少,个体加权残差的分布亦将趋近于零,称之为残差变异的收缩(ε-shrinkage) [35-36]。
个体间变异和残差变异的收缩值在一定程度上反映了诊断图的可信程度,收缩值越大,则诊断图越不可信。
对某个参数基于经验贝叶斯的诊断依赖于其个体间变异。
个体参数可以结合参数先验分布、残差变异以及个体观测值,通过贝叶斯方法估算获得。
基于经验贝叶斯估算的模型诊断主要包括以下几个方面:
①个体间变异和残差变异的收缩值一般应小于20%[30];
②绘制参数和个体间变异的散点图矩阵(scatter matrix plot of parametersand ETAs) 考察各参数之间的相关性;
③个体间变异直方图和Q-Q 图(histogram and quantile-quantileplot of ETAs) 是检验参数变异正态性的一种有效手段;
④参数的个体间变异对协变量的诊断图常用来检验参数和协变量之间的相关性。
3.7.3.2 自举法
自举法主要包括重抽样生成自举数据集、用NONMEM 将待评价模型和自举数据集进行参数拟合,上述过程重复多次后对参数估算值进行汇总分析。
成功应用自举法应满足以下两个条件:
①每个自举参数的2.5% ~ 97.5%区间包含原始模型估算参数;
②满足预先设定的稳健率,如稳健率> 80%。对运行时间较长的复杂模型,自举法计算耗时太长,不宜使用。
对于小样本数据,自举法也不适用。
此时,可采用抽样重要性重抽样算法(sampling importance resampling,SIR)。
3.7.3.3 可视化预测检验
如果模型能准确描述原始数据特征,则基于模型产生的模拟数据应能再现原始数据的分布,包括数据的集中程度和离散趋势。
可视化预测检验(VPC) 依赖于图形化手段呈现模型预测值和观测值的相符程度,而数值预测检验(NPC) 则将结果转化为模拟数据与观测数据的统计比较[37]。
VPC 包括模拟数据集的生成、统计量的计算以及图形结果的呈现。
采用VPC 的过程中需要注意以下几点:
①VPC 模拟次数的多少取决于评价目的与研究的问题,比如说要评估中心趋势还是分布的拖尾现象。
②通常选择95% 或者90%分位数来呈现模拟数据与观测数据,但这在很大程度上取决于每个时间/时间段内的观测点数量。
③划分时间段的主要依据是将浓度值相近的点放到一个组内。分组过少,如将早期的观测点与后期的观测点放到一个组内,可能会导致组的跨度过大,掩盖了模型拟合程度方面的信息。
相反,分组过多会减少每个组内的观测点数量,可能会使模型表现变差。
④当数据集包含不同剂量水平或其他显著影响预测行为的协变量(例如,给药间隔,给药途径,基因型,肾功能等) 时,需要根据剂量或相关协变量因素分层绘制VPC 图形,以避免将由数据不平衡所致的预测性不佳,解读为模型错误。
⑤常规VPC 在划分时间段时,未考虑剂量和协变量的变异,且常规VPC 用于剂量适用性研究(如剂量调整) 时敏感性会降低,可采用预测校准VPC (prediction-corrected VPC,pc-VPC) [38] 进行评价。
另有学者提出了标准化VPC(standardizedvisual predictive check,SVPC) [39],根据受试者特征,对预测区间的计算进行标准化,以提高其识别结构模型的错误或随机效应估计不足的能力。
可视化预测检验结果中,观测数据与模拟数据的分布特征通常以图形化的方式呈现。
如图7所示,空心圆点代表原始观测数据,实线代表观测数据的第5、50 和95 百分位数,虚线代表基于模型模拟数据的第5、50 和95 百分位数,阴影区域代表模拟数据对应分位数的95% 置信区间。

由图7 可见,在中位数和第95 百分位数附近观测数据与模拟数据具有相似分布特征,而在第5 百分数附近分布差异较大。
3.7.3.4 数值预测检验
NPC 的原理与VPC 相似,但其结果呈现方式侧重于数值统计量的比较。NPC 通过基于目标模型的模拟数据,构建多个预测区间(例如0、20%、40%、50%、60%、80%、90%、95%),然后统计观测数据在预测区间之外的计数或比例,并与期望值作比较。
NPC 可同时在多个预测区间的水平上评估模型设定,与仅能同时考查1 个预测区间的VPC相比,NPC 在某种程度上可提供更多的信息。
此外,由于NPC 分别比较每一个观测值和对应模拟值的分布,故无需进行剂量归一化和确定“时间段”。
然而,NPC 评价过程中未考虑时间维度的影响,因此可能无法识别随时间而变化的高估或低估趋势[40]。
3.7.3.5 敏感性分析
敏感性分析(sensitivityanalysis) 是一种定量描述模型输入变量对模型输出的重要性程度的方法[41]。
PopPK/PD 研究实践中,应该对不确定性因素如异常数据、模型假设、结构或随机变异参数等进行敏感性分析。
如果模型结果对不确定因素敏感,比如清除率对异常数据敏感,这时需要谨慎对待数据和模型,如果不是数据质量问题,是否存在模型没有描述的亚人群等问题。
敏感性分析的结果需要临床团队共同讨论而后通过图形、表格与数字形式呈现在报告中。
3.8 推断与模拟
最终模型确定后,分析人员需要对模型及参数进行解读。
基于模型参数的置信区间(confidence interval,CI),可以推断协变量对于参数的影响:
(1) 如果置信区间包括无效值如0,并且参数能够精确估算,表明数据不支持此协变量。
(2) 如果置信区间足够窄并且不包含无效值,表明数据支持此协变量。
(3) 如果置信区间很宽并且不包含无效值,表明数据不足以精确估算此协变量。
分析者必须根据临床或参数本身,自己定义“足够窄”和“很宽”的含义。
基于最终PopPK/PD模型进行推断,需要考虑协变量在模型中估算的精度以及协变量在临床中的实际影响。
即最终PopPK/PD 模型得出的推论应当包含模型参数的精度与变异,如相对标准误差(relative standarderror,%RSE)、95% 置信区间以表征模型预测值或模型参数不确定性的范围等。
PopPK/PD 模型的模拟可以提供固定效应、个体间变异及相关性、残差、参数不确定性等多个层级信息的预测。
常用的模拟层级:
①基于固定效应,例如典型受试者PK/PD 曲线模拟;
②基于固定效应及参数不确定性,例如协变量作用的森林图;
③基于固定效应、个体间变异及相关性、残差,例如PK/PD 曲线及分布区间模拟。
模拟引入的信息量取决于利用PopPK/PD 模型所要回答的问题,并不是模拟引入的信息越多越好。
4
PopPK/PD 模型分析质量控制与报告的一般考虑
4.1 质量控制
良好的质量控制(quality control,QC) 可保证准确科学地解决专业问题,同时确保规范的保存和记录数据、代码和结果,使第三方可重复整个分析过程[42]。
分析质量控制总体上包括验证(verification)和确认(validation) 两个相补过程。
验证包含程序是否正确编辑没有错误,输入的数据、NONMEM控制文件和输出文件对应关系正确。
确认判断模型是否能充分描述观察的数据,模型的确认(validation)可参考本文相关章节。
可信的模型需要验证和确认两个部分。
一般分阶段对分析进行QC。
以下列出大体的QC 环节和简要的内容。
每个分析单位应对QC 的内容进行细化。
同时建议采用基于风险的方法来确定需要QC 的内容。
例如可以按分析中发生错误的可能性、发现错误的难易度、错误对结果的影响对风险进行分级。
在任何QC 环节中发现了错误,则在修正后应该部分或全部地重新QC。
重复此过程,直到所有QC 检查通过。
通常情况下,QC 人员应独立于分析人员。
QC 流程一般如下:
(1) 首先对分析源数据进行QC,检查其中的错误、判断数据的完整性等。
(2) 对NONMEM 分析数据集进行QC,确认数据集的格式、数据是否正确。
当有些数据集包含的数据量大,在一定时间和人力的要求下,无法进行100% 的数据QC,则应进行风险评估,判断是否可以对部分受试者数据进行完整的QC。
(3) 在完成模型开发后,应对NONMEM 的控制文件、输出文件进行QC。
在此阶段,应对分析过程或内容进行审查,确保分析过程是根据数据分析计划执行的,如有必要,应再次确认项目组各方可接受分析计划的规定。
在模型开发过程中,可能涉及数百个模型,但由于时间限制,通常不可能对每个模型进行QC。
因此QC 一般仅关注报告正文中具体讨论的模型,至少包括基础模型和最终模型。
(4) 在完成以上内容后,进行分析报告质量和内容的QC。
该阶段包含对NONMEM 结果文件的图、表、列表的分析结果的QC。
如果分析的内容和报告需要向监管机构提交,则需要对数据和报告是否满足监管机构法规要求进行QC。
对报告的质量和内容进行同行评审往往很有必要,
可以对报告的总体研究思路、分析的正确性进行评估。
因为各有侧重,所以这个部分可以有多个不同背景的QC 人员,如专业QC、医学人员、具有建模经验的人员,从各个方面对报告进行全面QC。
严谨、美观、设计合理的报告,可以提高模型的可行度,增加可读性。
4.2 分析报告
分析报告通常包括9 个部分: 摘要、研究背景、目的、数据、分析方法与假设、结果、讨论、结论、附录。
以下对各部分进行描述[43]:
(1) 摘要: 摘要是报告中最重要的部分,应使用简洁易懂的语言描述。
作为概述,应包含主要研究背景,研究目的,主要假设,大体研究设计,数据和方法概况,重要结果,结论。
对决策有重要影响的图表、理由也可呈现在摘要中。
(2) 研究背景: 围绕研究的目的,阐述待分析药物特点,分析意图的背景情况、开展PopPK/PD分析的意义。
(3) 目的: 明确描述研究的目的,含主要和次要目的。如和方案不一致,则需说明。
常见目的有: 分析协变量对PK/PD 的影响,通过模拟选择合理的剂量等。
(4) 数据: 描述进行PopPK/PD 分析的临床研究,如研究设计、样本量(如受试者例数和血药浓度样本量)、采血时间点、受试者情况、给药信息(如药物名称、剂量、给药间隔、依从性)、药物浓度检测方法含定量下限、药效评价方法。
详细的人口学统计描述,使用图表探索协变量的特征和相关性。描述衍生变量的计算方法、数据格式、质控和数据整理的程序。如有不同分析数据集,也需描述它们之间的差异。
特别是描述PopPK/PD 模型分析不纳入的可用数据以及原因。(5) 分析方法与假设: 该部分应该提供详细的细节以达到可重现的目的。
详述各类缺失值的处理、低于定量下限数据的处理、异常值的鉴定和处理。
描述拟合算法(如贝叶斯、非参最大似然、FOCE),涉及的假设(如参数的分布)、模型构建的标准、使用的软件和版本。
详细说明结构模型(图解、公式),协变量模型,协变量筛选的方法和标准(如OFV、临床相关性),个体间、残差以及时间相关的随机模型,图法或统计方法模型评价的方法,还包括相关敏感性分析。
对于复杂的分析建议包含分析流程。
模拟方案的详细描述,包括虚拟人群的产生方法等和标准操作的偏倚。
(6) 结果: 一般包括结构模型、协变量筛选过程中的关键步骤和评价,最终模型的形式,最终模型的参数估计值以及变异(CV%)、精度(RSE%、95%CI),模型的评价,模拟或其他应用(如重要协变量对PK 暴露的影响、剂量的调整等)。
其中重点是最终模型的描述和评价。
分析的结果通常使用图表形式表达,图表中各种符号需清晰标注,便于理解,有时可选择彩色图进行指标区分。
结果的重点应围绕研究的目的和模型的应用,其他内容可以在附录中呈现,但并不意味着附录中的内容可以降低要求。
(7) 讨论: 讨论不是结果的重复,其主要是解释建模的结果,并在已有研究的基础上解释模型结果的临床意义,围绕研究目的评价模型解决问题的能力,包括用于建模数据的充分性和限制性,建模方法、假设验证的原理,对模型结果(如协变量、协变量对暴露) 的解释,其他研究对结果的支持(如类似药物研究),以及和常规单个临床药理研究的差异。
模型结果对给药方案的选择也可呈现在讨论中,如体重、肌酐清除对药物清除的影响,特殊人群给药方案的调整。
(8) 结论: 使用简洁易懂的语言,对模型的主要结果进行描述。
包括分析结果能解决的问题,如对说明书的支持、剂量调整方案以及临床相关性。
(9) 附录: 分析数据库的样式; 基础和最终模型的代码、输出结果; 分析步骤; 重要的图和表; 关键图形的绘制方法和代码等。
5
新药研发中的量效关系研究
量效关系指的是药物剂量、暴露量和临床效应(包括安全性和有效性等方面) 之间的关系,可以结合药理学基本原理,采用数学和统计方法进行探索和研究。
量效关系是PopPK/PD 研究的一个重要研究目的。
同时,PopPK/PD 是研究和探索量效关系中较为科学与普遍应用的方法。
量效关系研究是新药研发中不可或缺的重要组成部分。
了解药物的量效关系,对于评价药物的安全性和有效性、选择合适的给药剂量、调整给药方案、制定患者个体化用药方案等均具有十分重要的意义。
换句话说,量效关系研究是找到某新药分子能够被监管部门接受和最终批准的获益风险比的最佳患者人群、最佳用法用量(最佳给药剂量、频率、周期、时间等) 的核心研究。
5.1 量效关系的研究方法
5.1.1 前瞻性研究
一般情况下,药物的量效关系信息最好通过专门设计的对不同剂量进行对比的研究获得。
常用的研究设计包括平行研究设计、交叉研究设计、剂量滴定研究设计等,具体研究方案需根据药物特性、疾病特征和适应症人群等情况进行综合判断。研究设计可参考ICH 和FDA 等国内外相关技术指导原则。
为得到可靠的研究结果,复杂的研究设计需遵循统计学原理。
需注意的是,研究设计的重点应是阐明药物的量效关系函数,而非简单地进行不同剂量间疗效和安全性的成对比较。
若对量效关系曲线上某一特定点存在争议,例如对一个较低剂量是否有效存在争议,则可针对该问题开展单独的成对比较研究。
5.1.2 回顾性研究
某些情况下,也可以采用回顾性的分析方法,从固定剂量试验中获得不同浓度或不同暴露量下的效应数据,采用合适的统计方法进行分析,获得一定范围内血药浓度或暴露量与效应的关系。
但是,回顾性分析需关注不同受试者之间因疾病严重程度或其他患者因素导致的变异情况,需关注组间均衡性问题,应尽可能明确导致个体间药动学变异的因素,如人口学特征(年龄、性别、种族等)、肝/肾功能、饮食、合并用药等。
分析和解读研究结果时,应基于上述问题进行结论可靠性的判断,以及评估对指导后续试验设计等方面的参考价值有多大。
可采用PK-PD模型化或其他合适的统计方法进行量效关系分析。
新药临床研究结束后,还应对整个数据库进行回顾性分析,充分研究和挖掘可能的量效关系信息。
5.2 量效关系的研究变量
5.2.1 药物暴露变量的选择
一般情况下,药物或其活性代谢产物的体内暴露量常采用药时曲线下面积AUC、峰浓度Cmax、多次给药后的谷浓度Cmin等人体药代动力学参数为表征。
量效关系研究中,暴露量参数的选择通常需根据研究目的和研究设计等进行综合考虑。
药时曲线下面积AUC 是衡量体内药物暴露的经典指标,也是量效关系研究中最常用的暴露量参数。
多次给药达稳态后的AUC 对于评价暴露量与药物的长期效应具有重要意义。
量效关系研究中,峰浓度Cmax通常可用于评价与药物不良反应的相关性。
但Cmax受试验设计、采样时间、个体间变异等的影响较大,若采用Cmax评价量效关系,需进行良好的试验设计和采血点设计,以尽量获取真实可靠的Cmax数据。对于需长时间用药的情况,特别是慢性疾病的治疗,多次给药达稳态后的谷浓度Cmin一般与AUC 具有比例关系,此时可采用Cmin作为药物暴露量参数,进行量效关系分析。
临床试验过程中收集Cmin数据,相对于收集稳态AUC 数据,可在一定程度上减少采血点数量。
药物浓度是评价药物体内暴露最直接的变量,若在连续两次给药的间隔内,药物效应随时间可能发生变化或波动,则可以考虑直接采用药物浓度数据评价量效关系。
基于药物浓度-时间数据进行的量效关系分析可获得相对详细的时间依赖性信息,而这些信息一般很难通过基于AUC 或Cmin的量效关系研究获得。
对于某些特殊情况,还可以采用稀疏采点的血药浓度数据,进行群体药代动力学分析和贝叶斯估计,近似模拟群体或个体PK 暴露量参数,然后进行量效关系分析。
在难以获得完整PK 信息时,如儿童和老年人群研究,该方法对于评估量效关系具有重要作用。
5.2.2 药物效应指标的选择
新药的量效关系分析中,药物效应通常包括有效性和安全性等方面。效应指标可以采用临床终点、替代终点、生物标志物等。
若采用生物标志物,需确保与药物效应具有相关性,应得到临床医生和监管部门的认可。
在新药研发的不同阶段,量效关系研究所采用的效应指标很可能不同。
采用不同终点指标获得的量效关系也可能存在差异,此时应进行综合分析。
多数情况下,采用多个与临床终点具有相关性的效应指标进行量效关系分析,相对于仅采用一个指标进行分析的情况,可能获得更多有价值的信息,对于指导后续试验设计等问题可能更可靠。
5.3 早期量效关系研究
新药研发者应尽可能在临床试验早期进行量效关系探索,且同时关注有效性和安全性等多方面问题,这样可以在更早期获得更接近最佳剂量的获益风险研究数据,为后续预期和非预期的数据分析提供更加直接和可靠的数据基础,同时可以更早期探索到接近最佳获益风险比的剂量窗,可以在某些特殊情况下为撰写说明书中如肝功能损伤、肾功能损伤等特殊人群的用法用量时提供安全有效性评价基础(如实际上并未获得该特殊人群的直接安全有效性研究数据时)。
实际上,应在新药研发的整个过程中不断进行量效关系的分析和评价,比如在关键注册临床研究中若也同时采用多个研究剂量,将为上市后最佳剂量调整提供重要依据。
尽早进行量效关系研究,可在一定程度上降低后续临床试验的失败率,避免在后续更大样本量临床研究中收集到大量无效剂量或过高剂量下的临床研究数据。
尽早进行量效关系研究,最终可以提高关键临床研究中所选剂量为最优获益风险比的剂量的可能性,从而提高新药研发上市的成功率,减少因剂量问题导致的关键临床研究失败或未通过监管部门批准。
5.4 量效关系研究的一般考虑
对于新药而言,暴露量与安全性/有效性之间的关系是评估药物在某患者人群获益风险比的基本且重要的基础原理性信息,在指导新药研发和监管决策中均具有关键作用。
应注意有些药物虽然经过大样本量临床研究,但因为有效性或/和安全性原因未最终获得监管部门批准上市,其中不排除可能是由于关键临床研究的研究剂量并不是最优获益风险比的剂量引起。
此外,新药分子注册上市的关键临床研究通常是基于某整体人群的阳性研究结果,而通过量效关系分析,结合个体患者量效关系曲线与上述整体人群量效关系曲线的关系,可以使个体患者的用药方式最优化,即对于个体患者而言能够获得最佳治疗效果,达到个体用药的目的。
研究者应保证其研究过程的质量控制,以确保研究结果的真实性和可靠性,这决定了根据量效关系所做的最佳剂量(临床最优获益风险比)决策的正确与否。
6
总结与展望
本文主要介绍了新药研发中PopPK/PD 研究及其分析过程、质量控制与报告中的一般考虑。
希望读者了解到在创新药物研发实践中如何从PopPK/PD 角度思考药物研发,如何具体应用PopPK/PD 方法到药物研发过程中并直至完成对企业和监管机构都有效且高质量的分析报告。
本文不再赘述PopPK/PD 及量效关系的具体案例,可参考NMPA 和FDA 专家发表的经典案例总结[44-45]。
关于PopPK/PD 数据分析的基础理论和方法,也可参阅国内已发表的专著[34]。
新药研发中的PopPK/PD 研究是贯穿整个临床研发过程的迭代式研究,并且其应用会随着研究者的偏好和能力而涉及到临床试验的各个方面。
目前,PopPK/PD 研究在疾病进展(diseaseprogression)、临床试验模拟(clinical trial simulation)、市场定位及定价分析、药物经济学等方面均有拓展应用。
从技术角度看,PopPK/PD 采用的非线性混合效应模型理论已经比较完善,但是随着利用先验信息的贝叶斯方法在临床研发中的不断推广,结合先验信息的PopPK/PD 应用也将更加广泛。
伴随着各类临床终点的定量方法不断改进与提高,PopPK/PD 研究将为更多疾病领域提供更精准的量化模型。
相关领域如生物样本的检测技术、人工智能/机器学习技术等的高速发展都会对PopPK/PD 研究甚至药物研发产生深远影响。
随着PopPK/PD 技术体系的不断完善,其在新药研发中将得到更广泛且深入的应用。
在我国定量药理学专家们的不懈努力下,群体方法目前已经有了一定的技术积累与人才储备。
但由于我国创新药物研发起步较晚,如何将其落实到新药研发的各个环节,如何利用此方法实现更高效的药物研发和更好的药物监管则需要学术界、制药行业和监管机构的共同推动。
期待群体方法的应用能够最终为患者带来更科学合理的创新药物。