
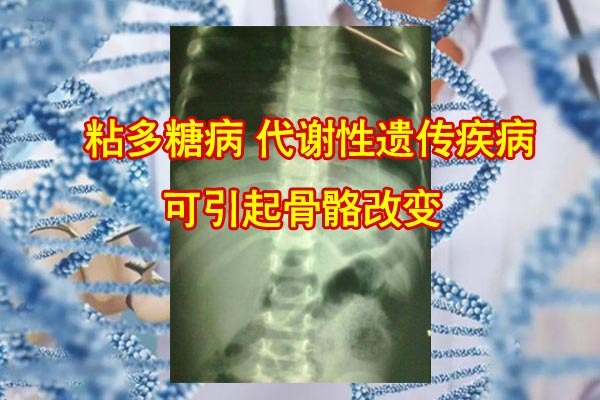

粘多糖病(MPS),也称粘多糖沉积症,是一种罕见的代谢性遗传疾病,其主要是因降解粘多糖(糖氨聚糖GAG)所需要的溶酶体水解酶的缺陷,导致组织内有大量的粘多糖蓄积,从而造成骨骼发育畸形、肝脾肿大、智力障碍和尿中粘多糖排出增多等症状。粘多糖病的总发病率约为1:250000,男性多于女性,多见于近亲结婚者的后代,多有家族史。粘多糖症无相应的特效药物,特异的治疗是骨髓移植。一般情况下,患儿多于出生1年后发病,10岁左右死亡,但有的病人可存活到50多岁。病变多以骨骼的病变为主,易导致脊柱侧弯。
发病机理
粘多糖实名为氨基葡聚糖,是骨基质和结缔组织细胞的主要成分,它是由糖醛酸和N-乙酰己糖胺或其硫酸酯组成的双糖单位的重复序列大分子,是多阴离子多聚体的糖胺多糖,其中的主要成分有硫酸皮肤素(DS)硫酸类肝素(HS),流散角质素(KS),硫酸软骨素(CS)和透明质酸(HA)等。这些多糖的降必须在溶酶体中进行,目前已知有10种溶酶体糖苷酶、硫酸酯酶和乙酰转移酶参与其降解过程,任何一种酶缺陷会造成氨基葡萄糖链的分解障碍面积聚体内并自尿中排出。
粘多糖病类型
粘多糖病可分为Ⅰ-Ⅶ等6型,其中V型已改称为IH/S型,每型又有若干亚型。以I型为多见,临床表现最典型。除Ⅱ型为X-连锁隐性遗传外,其余均为常染色体隐性遗传病。病变多以骨骼的病变为主,还可累及中枢神经系统、心血管系统以及肝、脾、关节、肌腱、皮肤等。
粘多糖病I型最常见
粘多糖病I型有2个亚型,均为α-1艾杜糖醛酸苷酶(α-Iduronidase)缺乏症,系因该酶的某种等位基因的突变所致。
粘多糖病I-H型(MPS-IH型),又称Hurler综合征,Hurler基因位于1号染色体上。在粘多糖中硫酸皮肤素和硫酸肝素中有L-艾杜糖醛酸的成分,其降解需要α-L-艾杜糖醛酸苷酶。由于此酶缺乏,其前体物的降解受阻而在体内堆积。硫酸皮肤素和硫酸肝素为角膜、软骨、骨骼、皮肤、筋膜、心瓣膜和血管结缔组织的结构成分,多为细胞膜外层的结构成分,细胞死亡后可释放出堆积的粘多糖。
粘多糖病Ⅱ型多心衰致死
粘多糖病Ⅱ型(unter syndrome)为X连锁隐性遗传。
病因是艾杜糖醛酸-2-硫酸酯酶缺乏。临床上有重型(A)和轻型(B)。由于酶缺乏使硫酸皮肤素(DS)和硫酸类肝素降解障碍,在体内储留并由尿中排出,二者的排出量比为1:1。
临床上重型表现与粘多糖I-H型相似,多在青春期前死亡。起病在2~6岁,有特殊面容和骨骼畸形,但脊椎无鸟嘴样畸形。角膜内皮细胞虽有粘多糖沉积而无角膜云翳,皮肤呈结节性增厚以上臂和胸部为著。幼儿期始有听力损伤,呈进行性耳聋,视网膜变性,心脏增大可闻收缩期与舒张期杂音。最后可发生充血性心力衰竭或心肌梗塞,常是死亡的原因。智能落后的差异较大或严重或轻度落后。肝脏肿大,和关节强直。轻型无智能障碍,临床症状较轻。
粘多糖病Ⅳ型易引起脊柱侧弯
粘多糖病Ⅳ型(Morquio氏病),有两个亚型。其病因为ⅣA为半乳糖-6-硫酸酯酶缺乏,ⅣB为β-D半乳糖酶缺乏。硫酸软骨素(CS)和硫酸角质素(KS)的降解障碍,而在细胞内沉积,硫酸角质素与软骨素-4/6-硫酸由尿中排出增多,但粘多糖总量不增多。随着年龄的增长硫酸角质素的浓度下降,至成年时排出量可正常。由于粘多糖在骨和软骨细胞沉积,骨发育障碍最为明显。Ⅳ型为常染色体隐性遗传。
Ⅳ型的临床特点为明显的生长迟缓,步态异常和骨骼畸形且逐渐显著。骨骼的畸形表现和I-S型相似,脊椎的鸟嘴突,椎骨扁平,飘带肋骨,还可有鸡胸,骨质疏松,髂骨外翻,股骨头变平,腕和膝关节肿大,但无关节强直。颜面呈颌骨突出,鼻矮,口大、牙间隙宽及牙釉质发育不良。学龄期出现角膜混浊,皮肤增厚且松弛。智力发育基本正常,青春期发育可正常。逐渐出现脊髓压迫症状,晚期出现麻痹性截瘫和呼吸麻痹。病人寿命多为20~30岁。
(东方红星/文/李烈;资料来源:百度文库,罕见病文献,航空总医院脊柱科,配图/网络)