
导 语
近年来,空间转录组(Spatially Resolved Transcriptoms, SRT)技术的进步使得利用组织中的空间信息进行基因表达谱分析成为可能。目前,关于SRT的实验策略大致可分为两类:第一类是具有单细胞分辨率的原位杂交或测序技术(包括seqFISH、MERFISH等),能够检测单个细胞中数百到数千个基因的表达水平;第二类是基于原位捕获的技术(包括SLIDE-seq和10x Visium等),即先进行空间条形码编码再进行测序,可以检测捕获特定位置中数千个基因的表达水平。这些不同的SRT技术使得揭示异质组织的复杂转录结构成为可能,并增强了人们对疾病细胞机制的理解。
在SRT研究中,一个重要的步骤是区分不同的组织区域,即定义在基因表达和组织学上空间一致的区域。传统的聚类方法,如K-means、Louvain等通常只能将基因表达数据作为输入,由于缺乏对空间信息和组织学的考虑,由此产生的聚类可能不具有整体性和连续性,不能反映真实的组织结构。
SRT研究中另一重要环节是鉴定出空间变异基因(Spatially Variable Genes, SVGs),将空间结构域与生物功能联系起来。已知的用于检测SVG的方法,如Trendsceek、SpatialDE和SPARK等只能独立地检测每个基因,并返回一个P值来表示基因的空间特异性,同样出于对空间结构考虑的缺乏,这些方法检测到的基因并不能保证特定的空间表达模式,因此难以利用这些基因进行后续深入的生物学研究。
为了同时解决上述两个难题,近日,美国宾夕法尼亚大学佩雷尔曼医学院生物统计系李明瑶教授课题组在Nature Methods期刊在线发表了题为“SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network”的文章,提出了空间转录组数据分析的新算法SpaGCN。SpaGCN首先构建表示数据空间依赖性的无向加权图,再将基因表达、空间位置和组织学进行整合来识别空间域。此外,SpaGCN还可以检测每个空间域丰富的SVG,通过将搜索空间限制在空间域上,以保证检测到的SVG都具有空间表达模式。SpaGCN适用于分析多种类型的SRT数据,包括ST、10x Visium、SLIDE-seqV2等。

文章发表在Nature Methods上
主要研究内容
SpaGCN算法工作原理
SpaGCN是一种利用图卷积网络分析空间转录组数据、划分不同组织区域并寻找区域富集基因的机器学习算法。SpaGCN首先通过构建一张加权无向图(Undirected Weighted Graph)将空间转录组中的基因表达和空间信息进行整合;随后利用一个图卷积层来聚集来自相邻点的基因表达信息,并把信息输送到一个分类层将整个组织划分成不同的区域;同时使用无监督迭代聚类算法对这些邻近点进行聚类并划分为特定空间域。
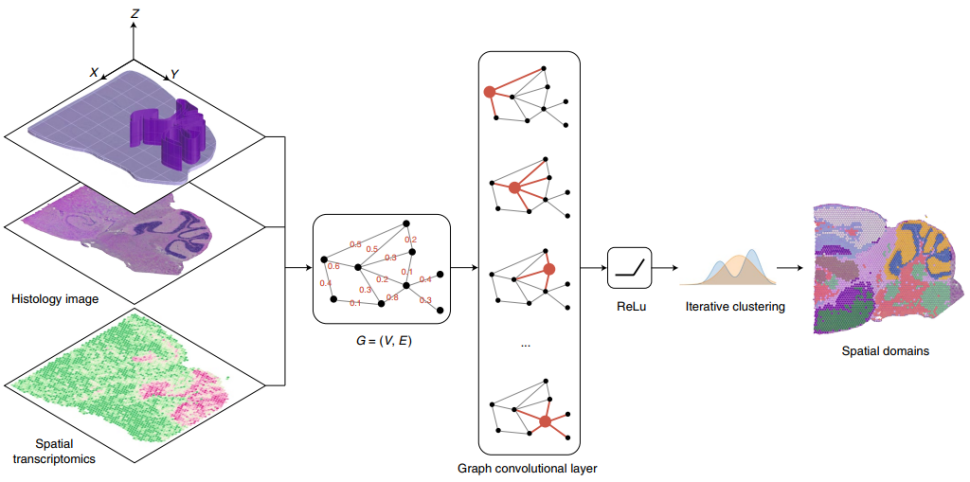
图1. SpaGCN算法工作原理概图,来源:Nature Methods
接下来,SpaGCN会在鉴定出来的空间域中寻找特异富集SVG,以确保找出的基因具有相似的表达模式,可用于进一步研究该组织区域的功能。当单个基因不能标记一个结构域的表达模式时,SpaGCN将构建一个由多个基因组合而成的复合基因来表示该结构域的表达模式。

图2. SpaGCN算法工作原理概图,来源:Nature Methods
利用SpaGCN分析人类原发性胰腺癌数据
为了证明纳入组织学信息的重要性,研究团队分析了使用ST技术生成的人类原发性胰腺癌数据,该数据集包括224个点和16,448个基因,以及三个手动注释的组织区域。组织学图像显示了癌区和非癌区之间的明显差异,表明组织学对聚类有参考价值,而SpaGCN具有对组织学建模的灵活性参数,该参数能够在检测每个点的邻域时赋予组织学的权重。通过默认值,SpaGCN即可很好地检测到非癌区域;当将组织学权重增加时,SpaGCN不仅能检测到非癌区域和癌区域,还能检测到与手动注释的癌症区域一致的区域。以上实验结果充分表明,SpaGCN将组织学信息纳入聚类中是有用且必要的。

图3. SpaGCN在检测空间域层面的性能比较,来源:Nature Methods
SpaGCN应用于人类背外侧前额叶皮质数据
为了定量显示SpaGCN在空间域检测方面优于Louvain、stLearn和BayesSpace等算法,研究团队分析了使用10x Visium生成的人类背外侧前额叶皮质数据,这项研究对三个人脑中的12个组织切片进行了测序,这些切片跨越六个神经元层和人类背外侧前额叶皮质中的白质。结果显示,SpaGCN和BayesSpace分析得到的空间域比Louvain更符合手动注释的组织分区;stLearn利用了组织学信息,但其性能与Louvain、SpaGCN和BayesSpace相比仍然较差。
为了进一步验证已识别的空间域,研究团队计算了每个域的SVG。尽管SPARK和SpatialDE检测到的SVG数量远大于SpaGCN,但这两种方法检测到的基因无法区分不同程度的空间表达变异。SpaGCN检测到的SVG的Moran's I值显著高于SpatialDE和SPARK。

图4. SpaGCN在检测空间域层面的性能比较,来源:Nature Methods
接下来,研究团队将SpaGCN、SpatialDE和SPARK检测到的SVG进一步分析发现,与SpaGCN相比后两者检测到的基因缺乏空间模式,证明了SpaGCN检测到的SVG的准确性和优势。此外,在单个基因难以鉴定的时候,SpaGCN能够找到特定领域的复合基因。

图5. SpaGCN在鉴定SVG层面的性能比较,来源:Nature Methods
结 语
研究团队推出了一种结合基因表达、空间定位和组织学的方法——SpaGCN,能够来模拟基因表达的空间依赖性,并用于识别空间区域和鉴定空间富集SVG。性能验证实验的结果一致表明,SpaGCN可以识别具有一致基因表达和组织学的空间域,并且检测到的SVG具有更清晰的空间表达模式和生物学功能相关性。随着空间转录组在生物医学研究中的重要性和日益普及,研究团队希望SpaGCN的诞生能对研究人员的大规模空间转录组数据分析提供帮助。
参考文献
1. Hu J, et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods. 2021 Nov;18(11):1342-1351.
2. Eng, C. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019).
3. Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-00935-2 (2021).