▉ 引言

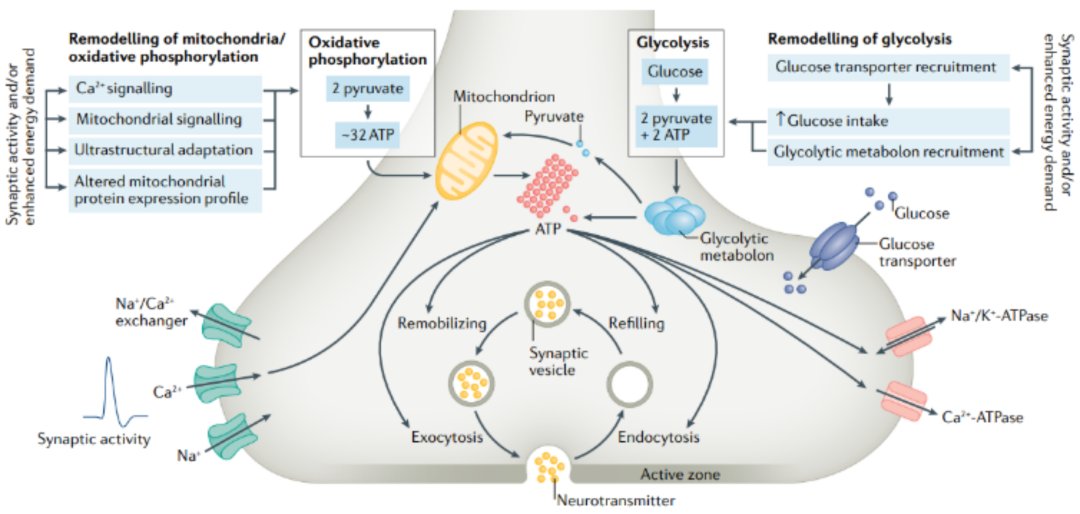
突触活动需要大量的能量,这些能量需要通过糖酵解和线粒体氧化磷酸化的局部三磷酸腺苷(ATP)合成来满足。ATP驱动动作电位,支持突触组装和重构,并促进突触囊泡填充和循环,从而维持突触传递。鉴于其极化形态特征——包括长轴突和末端区域的广泛分支——神经元在维持突触前能量稳态方面面临异常的挑战,特别是在密集的突触活动期间。
最近的研究已经开始揭示参与活动依赖和能量敏感的突触前能量调节的机制和信号通路或“突触能量代谢”(synaptoenergetics),这些概念上的进展已经建立了突触效能和可塑性的能量调节作为一个令人兴奋的研究领域,与一系列与生物能量衰竭和突触功能障碍相关的神经疾病有关。
今天为大家解读的综述是来自nature子刊的《Energy matters: presynaptic metabolism and the maintenance of synaptic transmission》。

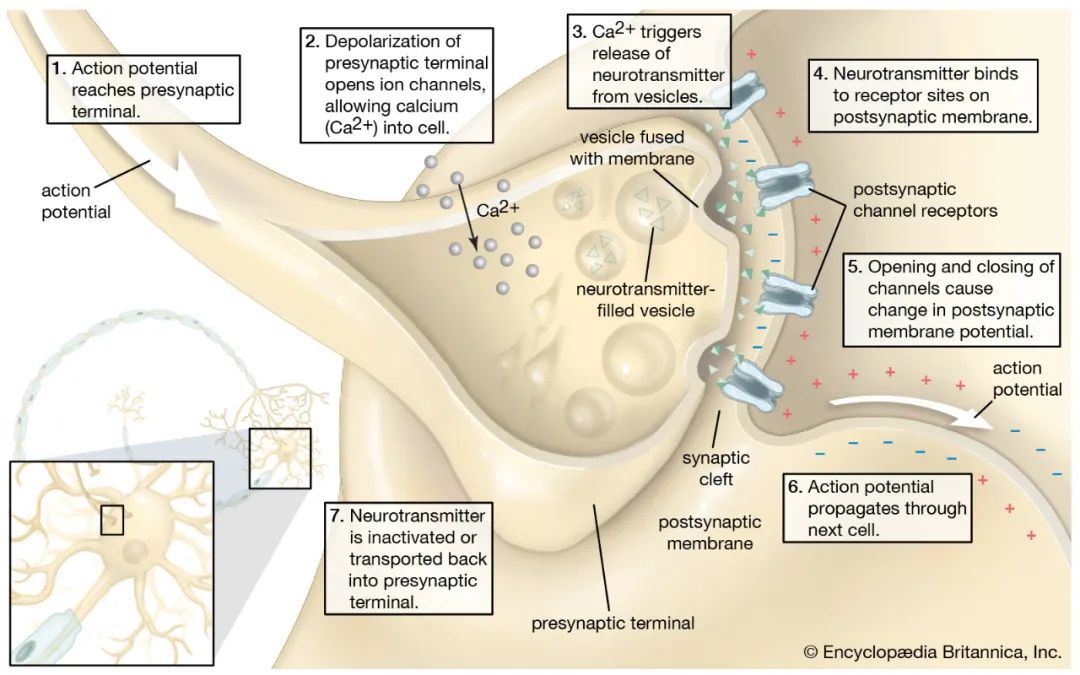
在人类的大脑中,大约有860亿个神经元连接在一起,形成了复杂的网络。在这些网络中,神经元通过突触传递相互交流,这种传递大约发生在1´ 105特定的细胞-细胞连接处,称为突触。突触传递开始于一个动作电位,去极性突触前膜和激活电压门控钙通道。这导致Ca2+迅速流入突触前末端,触发突触囊泡的胞外分泌和神经递质的释放。这些突触前事件产生了大量的能量需求,这些需求由局部能量供应以三磷酸腺苷(ATP)的形式来满足,ATP是通过糖酵解和线粒体氧化磷酸化产生的。ATP的消耗对于恢复动作电位下的离子梯度、突触内物质运输、突触组装和维持、突触前Ca2+泵出以及突触囊泡的补充和循环都是必需的,从而延长突触活动期间维持突触的传递。神经元是高度极化的细胞,具有单一的长轴突,最长可达数百厘米,因此它们的突触前末端远离细胞体。远端突触内的生物能代谢的维持是非常具有挑战性的,但也是至关重要的:生物能衰竭会显著影响突触传递的维持,以及一般的神经元功能。
在这篇综述集中讨论了神经元在高强度活动中保持突触前能量代谢以维持突触效能所面临的挑战。最近的进展提供了一系列令人兴奋的证据,说明突触前的能量供应是如何维持和调节的,以响应突触活动中增强的能量需求,突触前ATP稳态如何影响突触的效力和可塑性,在生理和病理条件下,能量不足是如何导致突触功能障碍的。由于篇幅有限,我们将讨论范围限制在解决这些关键问题及其影响的最新发现,重点关注谷氨酸突触。
如果想从不同的角度获得更多的见解,我们请读者参考其他关于“脑能量代谢”、“线粒体运输”、“质量控制和Ca2+信号”、“线粒体裂变和融合”、“突触前糖酵解和生物能衰竭”的深入综述。
▉突触能量概述
1. 突触是ATP消耗的主要部位
虽然成年人的大脑只占身体重量的2%,但它却消耗了人体总能量的20%。葡萄糖是生成ATP的主要碳源,其代谢过程中大约95%的ATP在大脑中产生。分析计算预测,神经元产生的总ATP中约有55%在轴突末端被消耗,主要用于为依赖ATP的膜泵(包括Na+/K+-ATP酶和Ca2+ -ATP酶)提供动力,这些膜泵重新分布离子梯度以维持静息电位。
突触囊泡循环也需要大量的能量消耗。据估计,每个谷氨酸突触囊泡循环需要消耗超过20,000个ATP分子,并且在突触前末端须有大约1´106个ATP分子用于恢复离子梯度和细胞内Ca2+水平。突触的活动可能会发生巨大的变化,因此在单个神经元和突触内的能量消耗会随着时间的推移而变化。例如,当神经元对强烈的突触刺激作出反应时,生物能需求就会急剧增加。因此,为了确保正确的信息处理,突触能量学必须以协调突触活动变化的方式被采用; 生物能学适应的缺陷将限制突触传递,从而削弱网络中的信息整合,导致认知功能障碍。
2. 通过糖酵解和氧化磷酸化生成ATP
当人脑处于安静状态时,糖酵解和线粒体氧化磷酸化的速率是非常匹配的:对于每一个葡萄糖分子通过 糖酵解 转化为两分子ATP和两分子丙酮酸,并促进氧化磷酸化生成 CO2、 H2O和30-36个ATP分子 。线粒体氧化磷酸化包括电子转移和ATP合酶的活性 (电子传递链的一部分) 。糖酵解和氧化磷酸化使葡萄糖完全氧化,导致氧与葡萄糖消耗的化学计量比接近6:1,并 提供了神经元使用的大约93%的ATP 。
然而,在神经元刺激的反应中, 能量代谢发生急性增加 ,以匹配 突触能量消耗的增加 。通过正 电子发射断层扫描 ,我们可以观察到当大脑活动增加时, 糖酵解会产生额外的ATP ,导致糖酵解速率和氧化磷酸化速率之间的短暂不匹配。神经元糖酵解是一种 非线粒体依赖 的 低量但可快速 生成ATP的途径 。糖酵解酶富集在神经元内的特定区域 (如快速轴突运输、突触囊泡和质膜上) ,在这些区域 运动ATP酶和离子转运ATP酶 需要快速供能。然而,考虑到神经元糖酵解的低量的产能, 仅通过这一途径产生的ATP不太可能为密集的突触传递提供足够的动力 。因此,对于 密集的突触活动 ,突触能量供应可能是通过 增加线粒体ATP的产生 和局部糖酵解来使能量最大化 。糖酵解与氧化磷酸化在刺激突触前膜功能中的贡献稍后将进一步讨论。
3. 星形胶质细胞可能为增强突触传递供能
星形胶质细胞可为增强突触传递提供能量。星形胶质细胞的代谢特征及其在 支持神经元生物能 方面的作用已经得到了很好的研究。已经提出,通过氧化磷酸化的葡萄糖氧化是神经元中主要的生物能量途径,而 糖酵解在星形胶质细胞中更为活跃 。在星形胶质细胞中,糖酵解可以 上调 ,以响应突触活动驱动的葡萄糖摄取增加,而丙酮酸进入三羧酸 (TCA) 循环和氧化磷酸化是一个限速步骤,因此不能上调; 相反,过量的 丙酮酸 被乳酸脱氢酶还原为乳酸。根据这一观点,人们提出了“ 星形胶质细胞-神经元乳酸穿梭假说 ”,即突触活动触发突触释放 谷氨酸 ,并将谷氨酸吸收到邻近的星形胶质细胞中,在星形胶质细胞上增加能量负担的过程,导致葡萄糖摄取和糖酵解代谢的增强,从而产生乳酸。然后星形胶质细胞向神经元输出乳酸,在那里转化为丙酮酸,为氧化磷化提供燃料。因此,星形胶质细胞-神经元乳酸穿梭假说 解释了神经元突触活动与星形胶质细胞糖酵解代谢之间的耦合 。
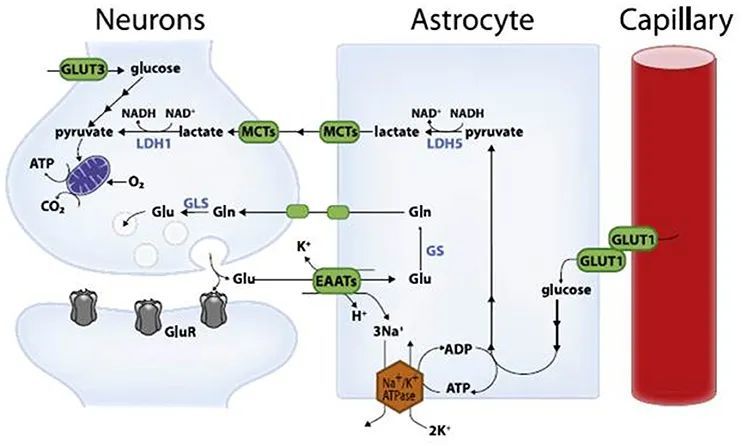
星形胶质细胞-神经元乳酸穿梭
事实上,星形胶质细胞具有独特的形态特征,非常适合 从血管中吸收葡萄糖,并将乳酸输送到邻近的突触 。虽然星形胶质细胞-神经元乳酸穿梭假说令人信服,但争论的一个关键问题是, 乳酸是突触激活后神经能量代谢的主要或唯一燃料 。此外,利用ATP生物传感器或代谢物标记,发现被激活的神经元主要 增强其对葡萄糖的利用,而不是对乳酸的利用 ,以进行能量代谢。符合这些发现,功能性老鼠大脑成像显示大脑活动直接与神经元中葡萄糖吸收和代谢有关。此外,在老鼠的花萼 (一个大型的听觉神经系统突触) ,在大脑切片中当突触受到刺激时,生理浓度葡萄糖是直接被使用。类似地,最近一项对脑切片的研究表明,单独提供乳酸不如提供葡萄糖有效,而且在海马网络中产生振荡的过程中甚至可能有害。我们测量了清醒小鼠海马切片和大脑中单个神经元的代谢反应,并观察到通过增强神经元的葡萄糖消耗,短暂地增加了胞质NADH/NAD+比值 (这是一种对葡萄糖代谢至关重要的氧化还原状态) ,而星形胶质细胞则不是这样。需要进一步的研究来确定 星形胶质细胞向神经元的定向流动是否可能在特定的亚细胞起作用 。研究胶质细胞产生的乳酸是否为基本的神经元活动提供能量,而不是增强突触传递也将是有趣的。
▉ 突触前的糖代谢
1. 突触前糖代谢活动增强
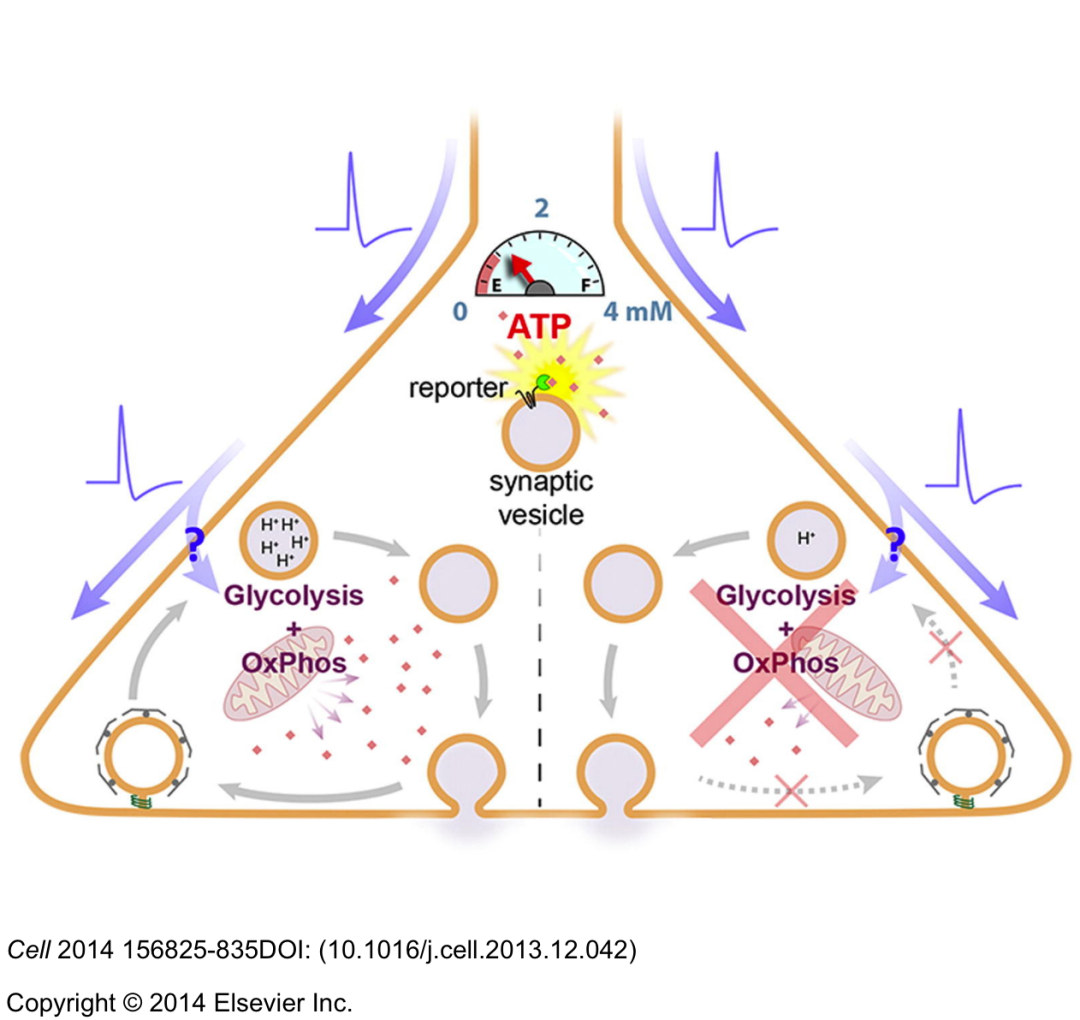
正如已经讨论过的,神经元糖酵解是一种 低产能 的途径,但可以提供快速的ATP供应,以维持突触前动作电位和突触基础传递。利用突触前靶向的 不休眠ATP报告基因“SYN-ATP” ,虽然突触活性通过糖酵解和氧化磷酸化刺激突触前ATP合成,但 选择性阻断糖酵解导致突触前ATP水平下降,并抑制突触囊泡循环 。这些发现表明 突触前ATP消耗是时空耦合的糖酵解机制 。
事实上, 膜结合糖酵解酶和离子泵 (包括Na+/K+-ATP酶,H+ -ATP酶和Ca2+ -ATP酶) 之间的联系已经被很好地证明了,拥有一个快速ATP供应可能是为这些离子泵提供动力的关键。这一假设得到了老鼠Held花萼的一项研究的支持,在该研究中, 糖酵解产生的ATP被证明是离子泵的燃料 ,在低频刺激下维持突触前动作电位。在果蝇突变体中,磷酸甘油酸激酶 (磷酸甘油酸激酶是糖酵解最后阶段生成ATP所需的一种酶) 的干扰降低了静止大脑中的ATP水平,并伴随着活动依赖性突触传递的丧失。这些研究表明糖酵解在维持突触活动所需的能量代谢中起重要作用。 糖酵解酶在突触前末端相对丰富 ,并在突触囊泡表面形成复合物,在那里它们产生ATP,为V型质子泵ATP酶和囊泡谷氨酸转运体 (VGLUT) 提供燃料。最近关于隐杆线虫的研究支持了 活性增强的突触前糖酵解 的概念,并显示了 糖酵解酶的重新分布 可能介导了这一过程: 在能量应激和神经元活动的反应中,酶会在突触前扣环附近积累,形成糖酵解代谢 (图1) 。干扰代谢的形成减少了ATP的供给,损害了突触囊泡的循环和恢复,改变了运动。
2. 葡萄糖在突触前末端吸收
考虑到突触前糖酵解酶的富集,我们需要考虑 葡萄糖如何到达突触前末端 ,以及 神经元是否适应在突触激活期间随着糖酵解的增加而增强葡萄糖摄取 。 GLUT3 (也被称为SLC2A3) 被认为是狗和大鼠神经元中主要的葡萄糖转运体,突触活动诱导其神经元表面表达的增加。 GLUT4 (也被称为SLC2A4) 表达在啮齿动物大脑的不同区域也有报道,GLUT4定位于细胞质中的储存器,并易位到细胞表面,以响应参与海马学习和记忆的胰岛素信号通路的激活。
最近的一项研究揭示了GLUT4动员到海马体神经末梢的新机制,其中 主能量应激传感器AMPK 被ATP供应不足和/或能量消耗增加所激活。AMPK代谢反馈将细胞内的GLUT4囊泡聚集到突触前膜表面,以促进葡萄糖摄取和糖酵解。此外,在持续动作电位放电期间, GLUT4的敲除抑制突触囊泡的循环 。有趣的是,这一过程与 运动诱导肌肉纤维中葡萄糖摄取 的过程相似。虽然 AMPK-GLUT4假说 很有吸引力,但还需要进一步的研究来确定GLUT4是否作为活跃的突触前末端的主要葡萄糖转运体。值得注意的是,增强葡萄糖摄取也可以通过提供更多的丙酮酸,以及通过 运输驱动蛋白结合蛋白1 (TRAK1,也称为Milton) 的糖基化,在 突触前末端阻断线粒体 ,从而促进突触前氧化磷酸化。
▉ 突触前线粒体代谢
1. 线粒体提供大部分突触前的能量
线粒体提供大部分突触前能量,水藤红素产生ATP的产量比单独糖酵解高得多,可提供总ATP的93%,而糖酵解提供神经元使用总ATP的7%。与星形胶质细胞相比,神经元含有更活跃的丙酮酸脱氢酶和更高水平的TCA循环活性,因此更倾向于氧化磷酸化而不是有氧糖酵解。
事实上,有证据表明,许多发生在突触前的主要能量需求过程都是由氧化磷酸化提供动力的(图1)。例如,通过刺激大鼠海马切片和氧电极记录相结合,研究发现,氧化磷酸化为神经递质释放提供了主要的能量来源。这表明线粒体来源的ATP对支持突触囊泡循环至关重要,而线粒体生物能量衰竭会损害突触前功能。
2. 突触前线粒体的生物发生、分布与维持
线粒体的生物发生、分布和质量控制对于维持远端轴突和突触线粒体,以支持突触前功能至关重要。然而,线粒体生物发生主要发生在细胞体中, 线粒体DNA 复制也发生在外周神经元的近端轴突和中枢神经系统神经元的远端轴突中。最近的研究表明,线粒体蛋白是在突触局部产生的,并与呼吸链复合物结合以维持线粒体功能。线粒体生物发生是由 PGCLA 控制的 (一个转录共激活子家族的成员,控制核编码线粒体基因的表达) 。新的轴突线粒体也可以通过分裂或裂变从原有的线粒体中产生,这一过程需要 GTPase动力蛋白相关蛋白1 (DRP1) 。通过 敲低TSG101 (线粒体生物发生的负调控因子) 激活PGCLA,可以 增加果蝇翅膀轴突线粒体密度和ATP生成能力 。这些表型可被DRPI缺失所抑制,支持 线粒体裂变参与轴突线粒体生物发生 的观点。
线粒体裂变和融合的竞争过程调节线粒体的大小、完整性、分布和能量代谢。携带DRPI突变的小鼠表现出突触形成异常和轴突线粒体分布缺陷。小鼠海马神经元中DRPI的缺失降低了线粒体衍生ATP的突触前维持,导致重复神经传递受损。同样,成年小鼠前脑神经元DRPI缺失降低了线粒体密度和突触前末端ATP的产生,从而破坏了高频刺激下的短期可塑性和突触传递。这些小鼠表型与DRPI突变的果蝇黑腹果蝇神经肌肉连接中观察到的相似,在那里线粒体局部化受损。线粒体大小的裂变依赖调节也在调节小鼠皮质锥体神经元突触生理中发挥重要作用。线粒体分裂因子(MFF)下调,MFF是位于线粒体上的一种DRPL受体。在这些神经元中,通过增强线粒体钙的摄取,增加线粒体大小,降低突触前Ca2+水平,从而导致神经递质释放受损。然而,在缺乏DRPI的小鼠Held终末的大花萼中,突触前线粒体异常聚集在一起: 这些突触中观察到的易释放池大小和突触囊泡循环的减少可能是线粒体生物能衰竭的原因。这些研究表明,有缺陷的线粒体裂变破坏了线粒体在轴突和突触中的分布,从而降低了突触前代谢。
线粒体融合需要三种大型GTPases的协调: OPAL, MFN1和MFN2。携带帕金森病(PD)相关的OPA1突变的多巴胺能神经元表现出轴突线粒体和多巴胺能突触的丢失。同样,腓骨肌菱缩症相关突变(MFN2)损害轴突线粒体运输,导致近轴突碎片线粒体聚集。这些研究表明,线粒体融合在维持轴突线粒体运输和分布中起着关键作用。
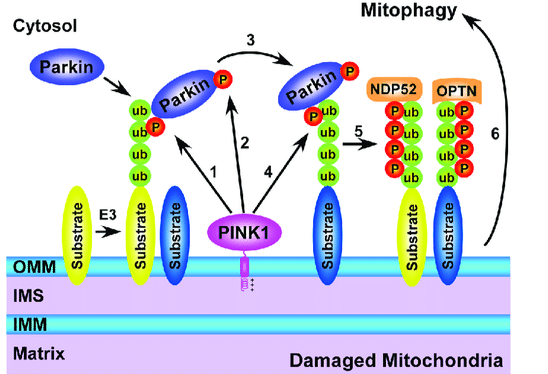
轴突和突触前线粒体通过逆行运输或局部线粒体吞噬被清除,当它们老化或功能失调。PINKI和parkin(两种PD相关蛋白)在激活线粒体自噬方面发挥关键作用。PINK1在线粒体外膜上积累,招募parkin去极化线粒体来激活自噬降解。在生理或慢性线粒体应激下,轴突线粒体向细胞体逆行通量降解,成熟溶酶体高度丰富。当轴突线粒体发生局灶性急性损伤时,PINK1-parkin通路被激活,局部线粒体自噬被诱导。有趣的是,与非神经元细胞类型相比,成熟神经元在线粒体沉积过程中表现出延迟的线粒体自噬反应,这表明在线粒体自噬被激活之前,神经元中已经存在早期的保护机制。当轴突线粒体遭受慢性和轻微的应激时,锚定在突触上的应激线粒体可以通过释放线粒体锚定突触蛋白(SNPH)向体细胞重新激活。总之,局部线粒体的生物发生、维持和质量控制机制维持了一个健康的轴突线粒体种群,从而保持突触前的能量供应。
3. 突触前线粒体维持密集的突触传递
在培养的小鼠神经元中,三分之一的 轴突线粒体 沿着轴突高度运动,从而有助于ATP在轴突中分散或扩散到突触前。然而, 锚定的突触前线粒体 处于一个理想的位置,可以充当“发电站”(图1)。而密集的突触活动迅速耗尽突触前ATP,这种局部能量不足可以通过将线粒体招募到突触前末端来逆转。使用早期版本的 ATP传感器 ,在培养的海马神经元中也观察到类似的活动诱导的突触前ATP水平下降。三维电子显微镜成像显示突触前线粒体在维持长期电位增强 (LTP) 中发挥关键作用。通过分析大鼠海马切片中θ脉冲诱导的LTP表明突触前活跃区中有33%的线粒体保留,这与培养的海马和皮层神经元的观察结果一致。
该研究进一步揭示, 线粒体靠近活跃区对突触前区大小和突触囊泡数量有强烈的正向影响 。在θ脉冲诱导的LTP诱导下,更多的突触囊泡从包含线粒体的突触前活性区储备池中被激活,从而维持突触效能。这些发现支持了先前对DRPI突变体的研究,在该突变体中,神经肌肉功能的线粒体定位失败导致突触囊泡从储备池中动员的缺陷。本研究还表明,抑制线粒体氧化磷酸化可阻断突触囊泡动员。因此,在密集的突触传递过程中,线粒体衍生的ATP在促进突触囊泡从储备池到释放位点的动员中发挥了主要作用。与此相一致的是,在Held的猫花萼和金鱼视网膜双极神经元的巨大突触中,需要 极高的能量需求 和 持续的ATP供应 来维持活跃和紧张的突触囊泡释放, 线粒体在突触前末端或活跃区表现出独特的聚集和积累 。
4. 线粒体生物能量表现的适应
如前所述,突触前线粒体非常适合 提供局部的ATP供应 ,以 维持密集的突触传递 。然而,一个相关的问题是,这些线粒体 是否经过活动依赖的重构 ,以适应其生物活性表现,以响应活动增强的能量需求。
在细胞信号水平上,有充分的证据表明, 突触活动增强了胞外钙离子流入突触前末端 ,线粒体通过外线粒体膜定位的 电压依赖性阴离子选择通道 (VDAC) 和 内线粒体膜定位的线粒体单分子通道 (MCU) 摄取过量的Ca 2+ 。线粒体 Ca2+的积累 不仅作为一个缓冲系统,而且作为 调节能量代谢的信号通路 。线粒体内钙离子的生理升高通过 激活氧化磷酸化作用刺激ATP合成 ,使线粒体内膜去极化,增加细胞呼吸, 激活依赖Ca2+的线粒体酶 ,从而 促进线粒体能量聚集 。 阻断线粒体Ca2+摄取 会阻止突触前ATP合成的加速,损害突触功能和可塑性。依赖钙的刺激向线粒体提供正反馈,以确保它们在延长的突触活动期间满足增强的能量消耗。一项对果蝇运动神经元终末的研究表明,线粒体在持续刺激过程中快速获得Ca 2+ ,Ca 2+ 进入刺激线粒体能量代谢,以协调突触前能量需求。
除了Ca 2+ 信号外, 抗凋亡蛋白BAD (也称为Bcl-xl) 在 改善能量代谢和维持突触可塑性 方面发挥着重要作用。在啮齿动物海马或皮层神经元中过表达BAD可以 招募突触前线粒体 ,增加线粒体生物量,并通过 减少质子泄漏 来增强生物能量能力。
在超微结构水平上,线粒体 嵴膜的表面积和密度与氧消耗和细胞色素氧化酶活性相关 ,因此,在非神经元组织中,线粒体嵴膜的表面积和密度是决定生物能量性能的关键超微结构参数。线粒体进行超微结构重构以响应增加的能量需求。在最近的一项研究中,在小鼠和人类大脑的GABAergic和谷氨酸能轴突中,检测了 突触前线粒体的超微结构 特征,揭示了 嵴膜密度增强 ,与低活性的突触相比,具有 较高突触活性的突触前线粒体的嵴层状结构和细胞色素C水平 。通过比较来自小鼠大脑的突触和非突触线粒体,两项研究揭示了与氧化应激和TCA循环相关的线粒体蛋白的独特表达模式。这些蛋白质的功能注释表明,突触前线粒体经历了一种适应,增强了能量代谢。
总之,这些 线粒体信号、超结构重塑和蛋白表达谱 表明,突触活性可以通过几种方式调节线粒体的生物能。
5. 突触前线粒体的招募和锚定
除了目前讨论的生物能适应外,活性依赖的线粒体 运输 和 锚定 调节能够确保代谢活性末端在持续活动期间有充足的ATP供应。
在神经元发育过程中,轴突经历动态 输出生长、分支和突触发生 ,因此需要更强的线粒体运输和更快的能量分散来为这些动态区域提供动力。然而,体外和体内研究表明,随着神经元的 成熟和衰老 , 轴突线粒体运动 在哺乳动物和非哺乳动物生物体中 下降 。在成熟神经元中,这种运动性的减少表明,在突触和轴突分支点上,固定的线粒体作为稳定的能量来源。线粒体在突触前位点的定位也增加了突触。在一项研究中,产生了用荧光蛋白靶向神经元线粒体的转基因有丝虫系,首次提供了成年小鼠神经系统线粒体运动减弱的体内证据。类似地,最近的两项体内研究报告了成年小鼠大脑中轴突线粒体的主要静态表型,可能是由于突触前位点捕获的线粒体数量增加。事实上,他的观点得到了对成熟神经元和成人大脑切片的多项研究的支持,其中大约30-50%的突触前活跃区保留着一个或多个线粒体。因此,这些研究表明, 神经元成熟是调节线粒体运输和锚定机制表达或修饰的主要信号触发因素 。
众所周知,线粒体的运输和分布受到调节,以响应生长状态、突触活性、衰老和病理应激的变化。 SNPH 是一个 静态锚定器 ,通过其羧基端跨膜结构域和氨基端轴突分选序列附着在轴突线粒体上,并通过其与微管的相互作用 保持线粒体静止 。相反,SNPH表达的升高会破坏轴突线粒体运输。轴突线粒体的移动和锚定是由马达驱动力和SNPH锚定力之间的平衡控制的。SNPH的表达随着神经元的成熟逐渐增加,并在整个成年期保持高水平。因此有助于在成年小鼠大脑中轴突线粒体的主要静态表型。
突触激活 可诱导局部突触前Ca 2+ 变化,不仅触发神经递质释放,还可 瞬时阻滞运动线粒体 ,从而使 线粒体重新分布到激活的突触 。 已经证明,密集的活动招募轴突线粒体,通过局部Ca 2+ 信号传导来维持突触前强度的稳态尺度,而这一事件通过表达MIRO1 Ca 2+ 不敏感形式而被消除。 MIRO1 Ca2+信号 是线粒体定位和稳态可塑性活性依赖调节的关键机制。然而,值得注意的是,在没有SNPH的情况下,MIRO1 Ca 2+ 信号可能不足以稳定地捕获突触前末端的线粒体。野生型神经元短暂的突触钙通道只能短暂地 (1s) 阻止线粒体运动3分钟。此外,在SNPH敲除的海马神经元中,强烈的突触活动未能阻止轴突线粒体。因此,假设SNPH通过协调MIRO1 Ca 2+ 信号转导,抑制KIF5 ATPase,这一 MIRO1 Ca2+-SNPH模型 与最近的光遗传学研究一致,该研究将轴突中的两种固定线粒体分类为“可移动的和不可移动的”。这暗示了不同机制下的线粒体瞬时阻滞与牢固锚定的MIRO1 Ca 2+ 感觉可能不是唯一的机制下的活动依赖的轴突线粒体阻滞。在使用MIRO1基因敲除小鼠时,据报道,MIRO1对Ca+调节的线粒体固定化不是必需的。另一项研究表明MIRO1缺失不会影响线粒体沿轴突的分布,这表明 其他机制可以弥补它的缺失 。
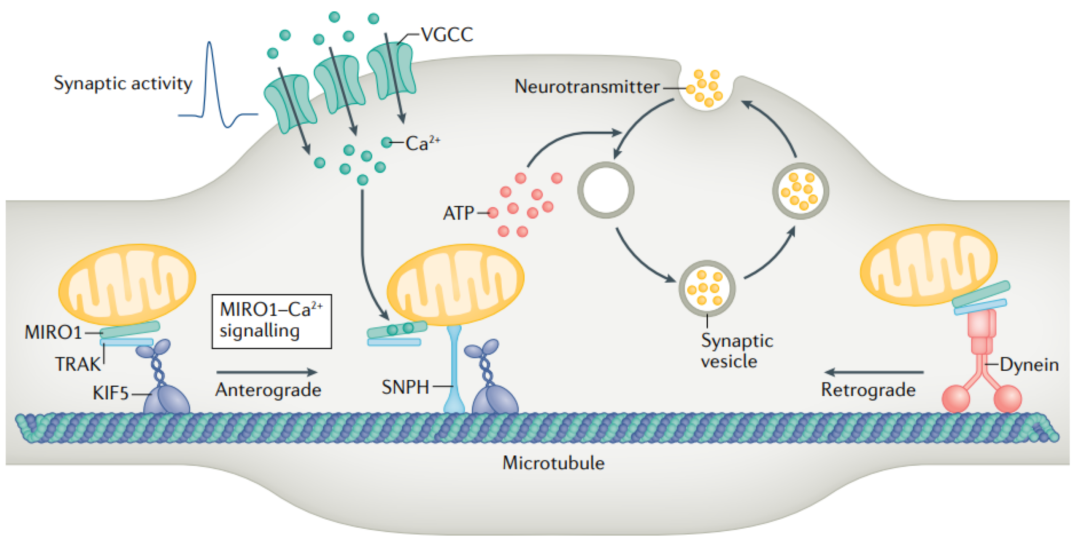
图2
运动适配器TRAKL和/或TRAK2 交替地将 KIF5 运动连接到线粒体,以独立于MIROI和/或MIRO2驱动向前运输。神经生长因子、神经递质5-HT和一氧化氮也被报道可固定线粒体。然而,目前还不清楚 这些途径是否有助于突触前线粒体锚定 。当驱动蛋白和动力蛋白马达驱动基于微管的长距离轴突运输时, 肌球蛋白马达介导沿丝状肌动蛋白 (F-肌动蛋白) 的短程运动。 F-肌动蛋白 在突触前末端形成密集的网状结构,从而成为 捕捉突触前线粒体的理想锚定平台 。SNPH敲掉的神经元突触前末端内的线粒体定位受损,表明轴突线粒体通过相互作用被募集到突触前末端并捕获微管转运和肌动蛋白锚定之间的关系。最近的一项研究表明, 肌球蛋白6 (MYO6) 驱动的线粒体补充和SNPH介导的线粒体锚定在突触前F-肌动蛋白上。SNPH作为一个适配器,招募MYO6到轴突线粒体;消耗MYO6或表达一种在SNPH结合中有缺陷的MYO6突变形式,可消除突触前末端对线粒体的捕获。因此, MYO6和SNPH之间的相互作用导致轴突线粒体从微管运输轨道切换到肌动蛋白锚定平台 (图3) 。也有报道称, MIRO1 通过在线粒体上 募集和稳定MYO19来介导线粒体-肌动蛋白的相互作用 。MYO6-SNPH和MYO19-MIRO1可能协同工作,以最大限度地维持突触前线粒体,或在不同的神经元或突触中独立工作。值得注意的是,Held的花萼可能有一个独特的机制来锚定与活性区相邻的线粒体簇,使用一个复杂的细胞骨架上层结构。
▉突触功能的能量控制
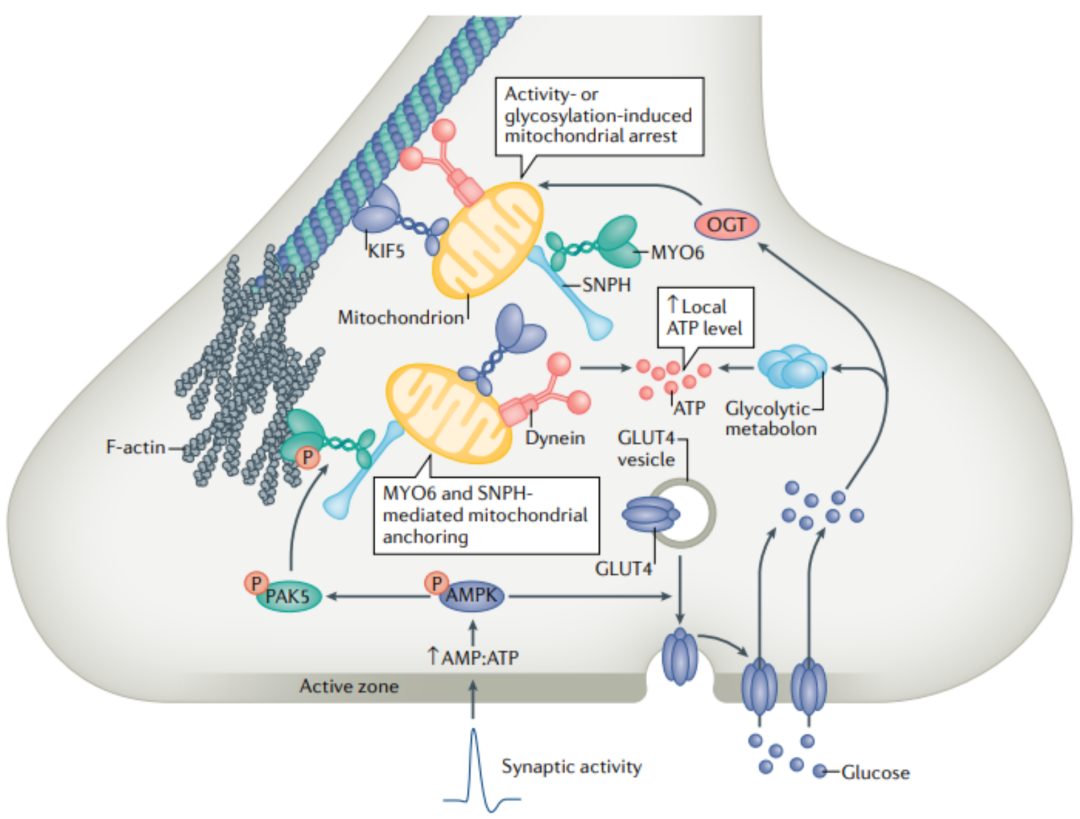
图3
1. 协调突触出现的调节
如前所述,神经元具有一种能量敏感机制,可以 招募和捕获突触前线粒体 ,以应对活动诱导的能量应激。 AMPK 是一种主要的 能量应激传感器 ,激活AMPK可同时 调节糖酵解和线粒体代谢 。研究表明,突触活动激活AMPK信号,进而适应糖酵解速率和线粒体呼吸,在高频刺激 (HFS) 中维持ITP。促进线粒体运输和突触前分布。失活AMPKA2, 催化AMPK亚基可以损害老鼠大脑海马LT P和认知能力。突触前能量应激可能激活AMPK信号级联。最近的一项研究解决了这一可能性,揭示了AMPK能量感应和SNPH锚定机制之间的关联。以前的工作表明突触前能源压力激活AMPK在远端轴突。 短暂快速脉冲刺激 导致蛋白激酶AMPK能量信号激活。此外, AMPK-PAK信号 轴在其运动域内磷酸化MYO6,从而增强MYO6驱动的突触前线粒体补充和SNPH介导的突触前F-肌动蛋白锚定。与MIRO1 Ca2+感知模型不同,该模型通过感知Ca 2+ 信号来瞬时阻止线粒体运输,而通过AMPK-PAK的能量感知使神经元能够牢牢捕获突触前线粒体。
因此,这两种信号通路可能首先通过感应 Ca2+瞬态 来协调 抑制临近突触微管上的运动线粒体 ,然后通过 感应活动诱导的能量应激 将 线粒体牢牢固定在突触前F-肌动蛋白 上。此外,如前所述,活动诱导的AMPK信号通过 将GLUT4招募到突触前表面 来 增强葡萄糖摄取和糖酵解 ,从而提供代谢反馈。AMPK信号通路可以刺激突触前糖解和线粒体氧化磷酸化,以协调突触能量的适应,以响应突触活性的增强 (图3) 。
2. 突触能量波动有助于突触变异性
突触传递在突触强度上显示出显著的 脉冲-脉冲变化 (PPV),这可能代表了电路中突触信号的计算优势,尽管在离子通道表达上存在差异, 突触囊泡释放动力学和受体浓度 都影响不同神经元之间和不同突触之间的突触强度变化。一项研究表明, 突触能量的波动 可能与小鼠海马神经元的PPV有关: 动态通过突触前模的移动线粒体引起局部ATP到二磷酸腺苷的广泛波动 (ADP比值通过影响ATP依赖的突触囊泡循环来诱导PPV) 。通过失活SNPH来增加轴突线粒体的运输,可以显著提高PPV,而通过过表达SNPH来固定轴突线粒体,可以消除PPV (图4a, b) 。在重复刺激过程中,在没有固定线粒体的情况下, 突触前末端ATP/ADP比率下降得更快,恢复得更慢 。本研究为中枢神经系统中能量调节突触前强度提供了新的见解。
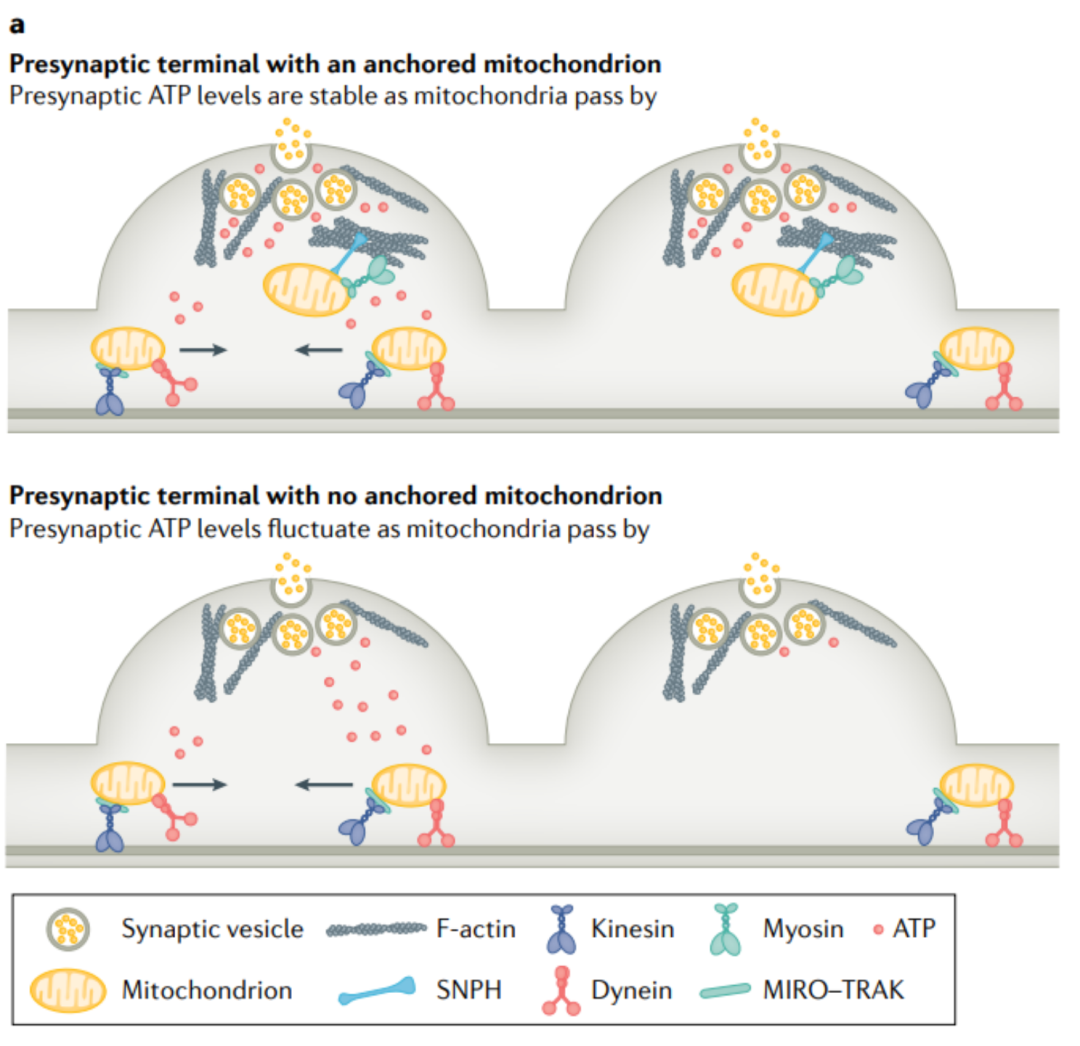
图4
3. 突触效能和可塑性的突触能量调节
线粒体在突触中发挥双重作用, 提供ATP 和 缓冲Ca2 +以维持突触传递 。Ca 2+ 从突触前线粒体的释放提高了细胞内钙水平,有助于短期或强直性强直后。一项关于Held花萼的研究表明,HFS期间线粒体Ca2+释放会提高突触前钙浓度在峰间间隔,以增强短期易化。 干扰能量信号 (AMPKA-PAK) 和 线粒体锚定 (MYO6-SNPH1) 之间的关联会损害突触前线粒体维护和局部Ca2+缓冲能力,因此,提高突触前Ca2+ 传递在重复活动和减缓短期突触前抑郁的作用。在小鼠听性脑干切片中研究了突触传递的能量调节,突触前花萼在活跃区附近持有一簇水线粒体,以维持高代谢率,以维持超过300 Hz的HFS。葡萄糖去除后突触前ATP的消耗加速了HFS期间的突触抑制和兴奋性突触后电流振幅的恢复受损。相反,当突触能量状态受到干扰时,突触前ATP会在几分钟的时间尺度内被HFS迅速耗尽。这种能量缺失在长时间活动后减缓了突触抑制的恢复,导致突触功效受损。另一项研究表明,在培养的大鼠交感神经细胞中表达一种功能缺失突变体syntabulin (一种线粒体KIF5运动适应子) ,减少了线粒体向轴突终末的运输,加速了突触抑制,减缓了HFS期间的恢复。与这一发现一致的是,在黑腹果蝇神经肌肉连接中,MIRO的消融会从突触前消耗线粒体,导致长期活动后的突触抑制。因此,突触前线粒体维持的 能量敏感调节 是 维持突触长期效能和可塑性 的重要机制。
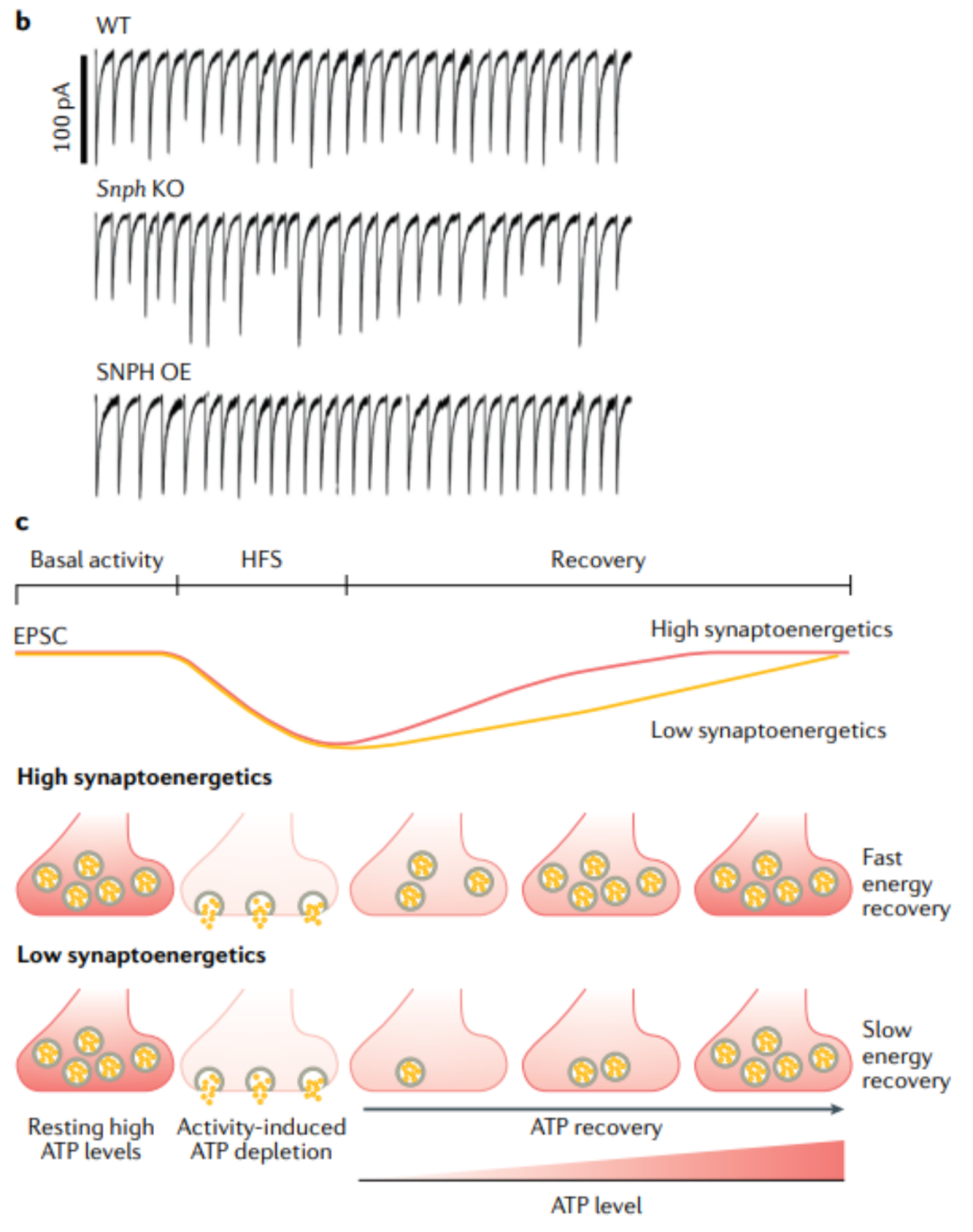
图5
4. 疾病中的突触能量衰竭
主要的神经退行性疾病的特征是生物能障碍,包括糖代谢受损和线粒体功能障碍,尽管在受神经退行性变影响的大脑区域观察到葡萄糖摄取和糖酵解的整体减少,监测疾病进展中的突触前糖酵解活性缺陷在技术上具有挑战性。线粒体ATP生成缺陷是发生在疾病进展早期的突触能量衰竭的主要原因,与突触功能障碍相关。在这里,我们讨论了三种主要神经退行性疾病的突触前线粒体功能障碍和生物能量衰竭的可用证据: PD、肌萎缩侧索硬化症(ALS)和阿尔茨海默病(AD)。
PD是一种神经退行性运动障碍,影响黑质多巴胺能神经元,表现为震颤、肌肉僵硬和运动缓慢。PD患者大脑中已观察到线粒体复合物I缺失,4个PD相关家族基因(PINKI、PRKN、PARK7和LRRK2)参与线粒体质量控制。对PINK 1基因敲除或PARK 7基因敲除大鼠纹状体突触线粒体的生物能量和蛋白质组学分析显示,线粒体呼吸功能受损,线粒体谱改变,蛋白质水减少平参与三羧酸循环和电子传递链受损的多巴胺释放多巴胺神经。然而,Lrrk2 -敲除小鼠的多巴胺能神经元在其神经末梢的呼吸链中显示出降低的蛋白质水平,当暴露于压力时,这些动物比野生型小鼠出现了更严重的运动障碍。突触前蛋白a-synuclein(α-突触核蛋白)是帕金森病的病理路易小体的主要组成部分,在氧化或代谢应激中诱导线粒体功能障碍,表达突变a-synuclein的转基因小鼠表现出线粒体变性和神经递质释放受损。事实上,在人类神经元中,a-synuclein水平的增加会破坏轴突线粒体运输,导致能量不足,导致突触变性。值得注意的是,在这些神经元中,MIRO1水平降低,而SNPH水平显著升高,反映了驱动力与锚定力之间的不平衡。对帕金森病死亡后幸存的多巴胺能神经元的分析显示,与对照组相比,电子传递链中的关键蛋白水平升高,剩余轴突和突触中的线粒体数量增加。这项研究表明,补偿性能量提升对多巴胺能神经元存活和多巴胺传递至关重要,强调了突触能量的拯救作为一种潜在的治疗方法。
▉ 结论
近年来的研究已经开始揭示突触能量代谢活性依赖和能量敏感调控的机制。突触传递的能量调节与一系列线粒体功能障碍、突触前线粒体维护受损、突触能量衰竭、突触功能障碍和突触丢失相关的神经系统疾病密切相关。糖酵解和氧化磷酸化对突触功能的相对贡献需要在有和没有固定线粒体的单个突触上进行检查。进一步的研究还需要了解突触活动和Ca2+信号是如何与能量传感通路协调,以促进神经元放电和长期突触可塑性过程中的局部能量代谢。
此外,尚不清楚活动是如何通过重塑线粒体的生物能量表现来诱导同步能量适应的。最近的一项研究揭示了一种外泌体介导的跨细胞途径,通过该途径,少突胶质细胞来源的乙酰化酶2被传递到轴突,通过线粒体蛋白去乙酰化增强生物能。该研究开辟了一个全新的研究领域,研究少突胶质细胞-轴突信号在能量代谢调节中的作用,超越了少突胶质细胞在电气绝缘中的作用。
在主要神经退行性疾病和神经障碍的早期阶段,生物能量衰竭是一个常见的问题,因此,它是否发挥了趋同的病理作用,导致突触功能障碍和轴突退化,以及挽救生物能量缺陷是否减缓进展或减弱发病机制成为紧迫的问题。了解突触能量失效背后的疾病相关机制对于设计能量拯救策略至关重要。
功能性磁共振成像和正电子发射断层扫描为脑代谢提供了有价值的见解。然而,目前的成像工具在评估突触水平分辨率的体内能量代谢方面能力有限,因此需要开发敏感ATP传感器来跟踪病变大脑中的突触能量动力学。这将有助于我们理解在健康、衰老和病理压力下,活动依赖的能量敏感途径如何调节突触前线粒体氧化磷酸化和糖酵解。新出现的证据表明,病变大脑中存活的神经元和突触的代偿能量代谢,为恢复或促进神经障碍早期阶段的突触能量提供了一种令人兴奋和有希望的治疗方法。
编辑人:宁帅同学