

邹 龙,刘荣丽 ,易 师
(湖南稀土金属材料研究院,长沙410126)
中图分类号:O657.31 文献标志码:B 文章编号:10014020(2017)07081404
钪作为稀土元素,具有独特的物理化学性质,如钪元素加到铝合金中,可以显著提高合金的再结晶温度,强烈抑制沿晶断裂倾向,提高合金强度、塑性和断裂韧性[12]。近年来,随着新材料技术的迅猛发展,氧化钪(Sc2O3)更是广泛应用于固体氧化物燃料电池(SOFCs)[3]、新一代激光晶体[4]、大功率金属卤素灯、高能辐射用核能屏蔽、特种陶瓷等领域。但氧化钪中含有的杂质元素会导致这些材料性能极大下降,因此人们对氧化钪的纯度要求越来越高,对所含杂质元素的控制也愈加重视,而铁是其中重要的杂质元素之一。
氧化钪中杂质铁的测定一般采用电弧粉末法[5]、电感耦合等离子体原子发射光谱(ICPAES)[6]、石墨炉原子吸收光谱法(GFAAS)[7]、分光光度法等。这些方法已成功应用于氧化钪中较高含量铁的测定,但当高纯氧化钪中铁含量小10μg·g-1时,上述方法受基体干扰严重,测定结果重现性低。针对高纯氧化钪中痕量非稀土杂质测定,电感耦合等离子体质谱法(ICPMS)被认为是最有效方法,但是对于杂质铁的测定,在氩气背景下存在的大量ArO+ 与铁的质子数重合(56Fe与38Ar18O/40Ar16O、57Fe与40Ar17O、58Fe与40Ar18O),严重干扰质谱测定。文献[8]采用动态反应池技术(DRCe),用甲烷替代氩气为反应气体,选择适当同位素及仪器工作参数,实现了DRCICPMS对高纯稀土中铁的测定,但是DRCICPMS运行成本高,维护费用高,制约了在实际生产中的应用。而研究高纯氧化钪中痕量铁富集分离,是提高测定结果的准确度和精密度的有效途径之一。目前国内外常采用的分离富集方法主要有离子交换法、萃取法和液膜法等。其中,固相萃取(SPE)由于具有使用方便、富集效率高、吸附剂选择灵活等特点,已发展成一种有效的多元素预富集方法[9]。本工作利用邻二氮菲与氧化钪中痕量铁可生成被活性炭吸附的配合物,使用纯化处理活性炭为吸附剂的固相萃取进行富集、洗脱,采用电感耦合等离子体原子发射光谱法测定富集后的铁,建立了测定高纯氧化钪中痕量铁方法。
1 试验部分
1.1 仪器与试剂
AgilentICPOES700型全谱直读电感耦合等离子体发射光谱仪;MilliQAcademic型超纯水仪;固液分离层叠式过滤器;聚四氟乙烯(PTFE)吸附
柱(50mm×9mm)。铁标准溶液:1.000g·L-1,称取高纯金属铁1.0000g,用硝酸(1+1)溶液30mL溶解,加热煮沸除去氮氧化物,冷却,移入1L容量瓶中,使用前用水稀释至所需质量浓度。抗坏血酸溶液:质量分数为4%。邻二氮菲溶液:质量分数为4%。缓冲 溶 液:称 取 六 次 甲 基 甲 胺 40g,加 水100mL,盐酸10mL溶解并摇匀。高 纯 氧 化 钪 模 拟 溶 液:称 取 高 纯 氧 化 钪20.0000g置于250mL烧杯中,加入盐酸50mL,低温加热溶解,并蒸发溶液至近干,加入水20mL冷却至室温,转移至250mL容量瓶中,用水稀释至刻度。AlfaAesar活 性 炭 为 分 析 纯,粒 径 0.38~0.83mm;盐酸、硝酸均为优级纯,使用前经亚沸蒸馏器蒸馏提纯;试验用水为超纯水。
1.2 仪器工作条件
发射功率1.0kW;等离子气流量15.0L·min-1,辅助气流量1.50L·min-1;雾化器压力200kPa;观测高度10mm,一次读数时间5s,仪器稳定延时15s,进样延时30s;泵速15r·min-1;清洗时间10s;铁分析谱线Fe259.940nm。
1.3 试验方法
称取活性炭500g,用纯化后的盐酸(1+1)溶液浸泡,固液分离层叠式过滤器过滤,多次水洗直到滤液呈中性,烘干备用。将PTFE吸附柱用盐酸(1+1)浸泡30min后,接入蠕动泵后以15r·min-1转速进水清洗10min,备用。
动态吸附:称取试样2.0000g置于250mL烧杯中,加入盐酸20mL,低温加热溶解,并蒸发溶液至近干,加入水20mL,冷却至室温,加入缓冲溶液将溶液pH 调至4.0~6.0,再依次加入抗坏血酸溶液2.5mL,邻二氮菲溶液5mL,转移至50mL容量瓶中,用水稀释至刻度,摇匀静置10min待测。将1 g 已纯化活性炭填充入PTFE 吸附柱(50mm×9 mm)中,接入蠕动泵后以20.0mL·min-1速率进水清洗5min,再以5.0mL·min-1速率进待测溶液。随后用硝酸(1+1)溶液10mL洗脱保留在柱上的金属离子,洗脱4min,将洗脱液转移至25 mL 容量瓶中定容,在仪器工作条件下测定。静态吸附:取50mLpH4.0~6.0的含不同质量浓度Fe(Ⅱ)的高纯氧化钪模拟溶液,依次加入抗坏血酸溶液10 mL,邻二氮菲溶液25 mL,振荡10min后,加入1g活性炭吸附铁的配合物,振荡30min,静置。移取上清液,用ICPAES测定上清液中铁的含量。
2 结果与讨论
2.1 酸度及配位剂用量的选择
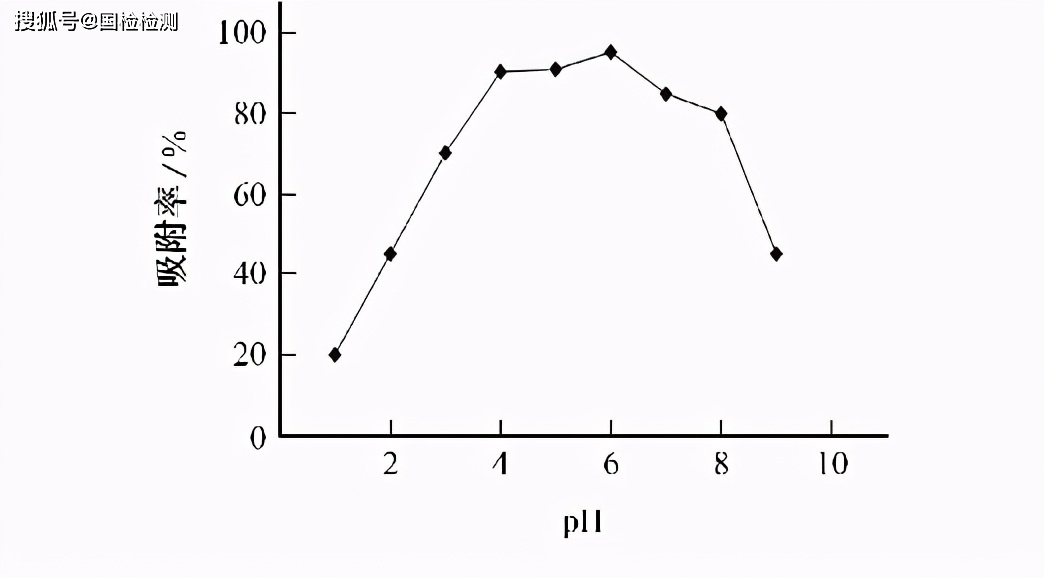
酸度是影响痕量铁分离富集的重要因素之一,配位剂邻二氮菲在酸度为pH2~9时才能与铁生成稳定配合物[10],而此配合物也只有在一定酸度下才能被活性炭吸附。试验将待测溶液调节至不同酸度,按试验方法测定,考察了pH 为1.0~9.0时氧化钪中铁的吸附率,结果见图1。由图1可知:当pH 小于4.0时,铁的吸附率随着pH 的增大而增大;当pH 为4.0~6.0时,铁的吸附率随着pH 的增大变化不大,说明铁可被稳定吸附;当pH 大于6.0时,铁的吸附率随着pH 的增大而减小。试验选择pH 为4.0~6.0。为保证加入的邻二氮菲能与溶液中铁配位反应完全,试验考察了不同邻二氮菲溶液加入量对铁的吸附率的影响,结果表明邻二氮菲用量在5mL左右时,吸附率大于95%且趋于稳定。试验选择配位剂邻二氮菲溶液用量为5mL。
2.2 活性炭用量及进样速率的选择
吸附剂活性炭作为吸附铁的载体,其用量是影响方法有效性的重要因素之一。活性炭的静态吸附容量是一个重要参数,决定了活性炭能最大限度吸附溶液中多少铁离子。固定溶液体积为50mL,活性炭用量为 1g,改变铁的初始质量浓度,按照1.3节中静态吸附试验方法进行考察,按公式(1)、(2)计算得活性炭对铁的吸附率和吸附量,结果见

式中:ρ0 为初始溶液中 Fe(Ⅱ)的质量浓度,mg·L-1;ρ为吸附后溶液中剩余的Fe(Ⅱ)的质量浓度,mg·L-1;犞 为试样溶液的体积,mL;犕 为活性炭的质量,mg;犃犱狊为吸附率,%;犙狊 为静态吸附容量,mg·g-1。
结果表明:在活性炭1g的用量下,当模拟溶液中的初始质量浓度超过4mg·L-1时,活性炭对Fe(Ⅱ)的 吸 附 率 显 著 下 降,计 算 可 知 活 性 炭 对Fe(Ⅱ)的最大静态吸附容量为4.84mg·g-1。
进样速率决定了样品溶液中铁离子与吸附剂活性炭接触时间的长短。进样速率过快,铁离子无法
被活性炭完全吸附,进样速率过慢则延长分析时间。因此,应选择合适的进样速率确保铁离子被快速高效的吸附。按照动态试验方法,将50mL样品以不同的速率进样,计算铁的回收率。结果表明:当进样速率在1.0~5.0mL·min-1时,铁离子的回收率均在90%以上;当进样速率大于5.0mL·min-1时,铁的 回 收 率 显 著 降 低。试 验 选 择 进 样 速 率 为5.0mL·min-1。
2.3 洗脱剂的选择
活性炭富集物的洗脱,一般选用盐酸或硝酸。按照试验条件考察了不同体积盐酸(1+1)溶液及硝酸(1+1)溶液以5mL·min-1的速率洗脱的效果,以铁的回收率为纵坐标,酸的用量为横坐标作图,其结果图3。
由图3可知:硝酸的洗脱效果优于盐酸,采用硝酸(1+1)溶液10mL能有效洗脱铁配合物,回收率在95%以上。试验选择10mL硝酸(1+1)溶液作为洗脱剂。
2.4 分析谱线的选择
用铁元素的标准溶液和空白溶液在各波长处进行扫描,得到各波长扫描轮廓图,然后输入洗脱液中的主要共存干扰元素钪、钙等标准溶液,得到相应的波长扫描图。根据计算机显示的这些谱线及背景轮廓和强度值,以及观测到干扰的类型和程度,选定铁的分析谱线为Fe259.940nm。
2.5 标准曲线及检出限
高纯氧化钪中痕量铁的线性范围为0.02~1.00mg·L-1,线 性 回 归 方 程 为 狔=1.134×103狓+1.465×10,相关系数为0.9995。检出限是用特定分析方法在给定的置信度内可从样品中定性检出待测物质的最低浓度或最小量[11]。按照国际理论与应用化学联合会(IUPAC)的规定,测定空白溶液11次,以3倍标准偏差计算得检出限(3σ)为0.052μg·g-1。
2.6 精密度和回收试验
取4种不同批次高纯氧化钪,每一批次样品分别准确称取12份,其中6份用于直接测定,并计算测定值相对标准偏差(RSD),另6份在溶样后加入4.00mL质量浓度为1.00mg·L-1的铁标准溶液(相当于样品中铁的加入量为2.00μg·g-1)进行加标回收试验,结果见表1。

由表1可知:回收率在71.0%~109%之间,RSD在2.0%~7.2%之间,方法准确可行。
本工作系统地研究了固相萃取活性炭分离富集电感耦合等离子体原子发射光谱法测定高纯氧化钪中痕量铁的试验条件,方法准确、可靠,能够满足当前科研生产的需要。