B细胞免疫耐受丧失是包括SLE在内多种自身免疫性疾病的免疫学基础。自身抗原通过toll 样受体(Toll-like receptors,TLRs)激活B细胞,破坏免疫耐受,产生自身抗体,进而有自身抗体及其自身抗体抗原复合物引起损伤。
B细胞的激活由四种受体控制,即结合自身抗原的B细胞受体(BCR)、细胞因子受体、参与同源T细胞-B细胞相互作用的受体(包括检查点分子)、以及包括TLRs在内的先天免疫受体。
TLR信号促进了B细胞介导自身免疫性疾病的三个关键机制:抗体的产生、向T细胞递呈抗原、及细胞因子的产生。
TLR7/TLR9的相反作用
TLR7在浆细胞树突细胞(PDC)、单核细胞和B细胞中以双等位基因表达,TLR7在女性这些细胞中的水平高于男性,促进SLE的发生。TLR9抑制TLR7的功能,保护机体免受于发生SLE。
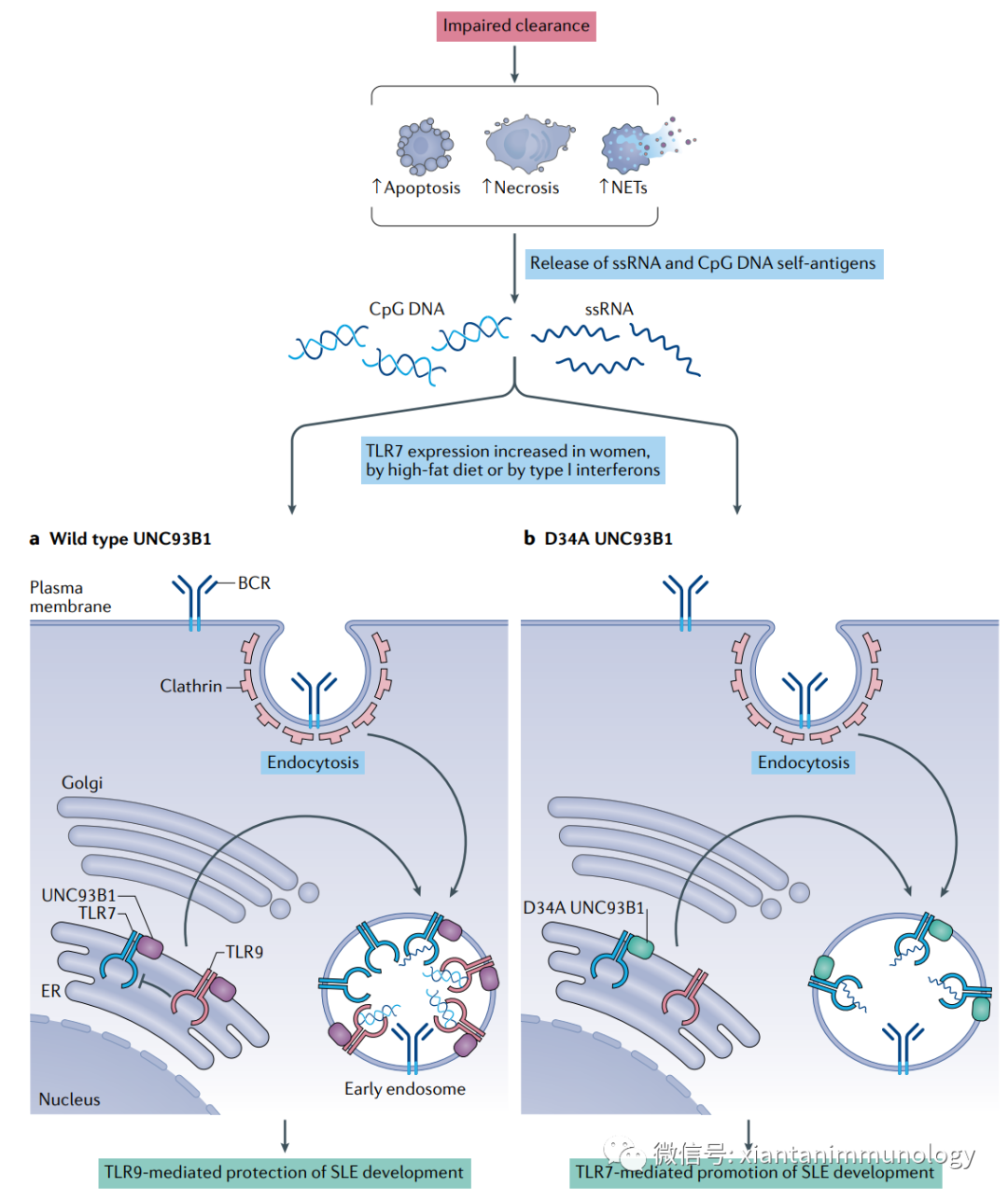
自身抗原通过中性粒细胞细胞外捕网(NETs),坏死细胞和凋亡细胞释放出来。这些自身抗原可以在B细胞表面被BCR识别,从而启动它们的细胞内化。一旦进入内皮体,自身抗原就可以在B细胞中触发TLR7和/或TLR9。
导致TLR7过度表达的基因或环境信号(如性别、饮食和细胞因子环境,包括I型干扰素的水平)增加了个体对SLE的敏感性。在生理条件下,TLR7信号受到TLR9的限制,它保护个体免受SLE的发展。
TLR9功能的破坏可以有利于TLR7信号传导,并促进SLE的发展。这种破坏可能涉及到细胞内蛋白UNC93B1,它推动TLR7和TLR9转运到内染色体隔室。跨膜UNC93B1中的D34A突变有利于其与TLR7的相互作用,可以在实验模型中提高内皮体中TLR7的丰度,增强TLR7信号,并启动致命的全身炎症综合征。
TLRs驱动浆细胞分化

系统性红斑狼疮(SLE)患者产生两种不同途径的致病性抗体分泌细胞:生发中心反应和滤泡外途径,这两种途径都与静息的初始B细胞有关。
生发中心通路产生双阴性B细胞(DN1B细胞;即IgD−CD27−CXCR5+),生发中心产生的记忆B细胞可以重新进入生发中心反应或分化为抗体分泌细胞,产生同型转换anti-Smith和anti-RNP。生发中心的自发产生取决于TLR7。
TLR7还驱动滤泡外途径,其中静息初始B细胞成为激活初始B细胞(CD11c+IgD+CD27−CD21−MTG+CD23−)随后产生IgD-CD27−双阴性B细胞亚群DN2B(IgD− CD27− CD11c+ Tbet+ CD69+ CD21− CD24− CD38− CXCR5− FCRL4− FCRL5+)。DN2B细胞是SLE患者致病抗体分泌细胞的前体,由TLR7、IL-21和IFNγ促进其分化。
值得注意的是,静息的初始B细胞也可以依赖于BCR的方式产生调节性血浆细胞,其特征是表达LAG-3。这些调节性血浆细胞对TLR信号反应,会产生高水平的IL-10。在稳定状态下,它们还分泌IgM,对受损细胞表达的抗原有反应性,这表明它们可能参与了受损细胞的清除。
药物开发
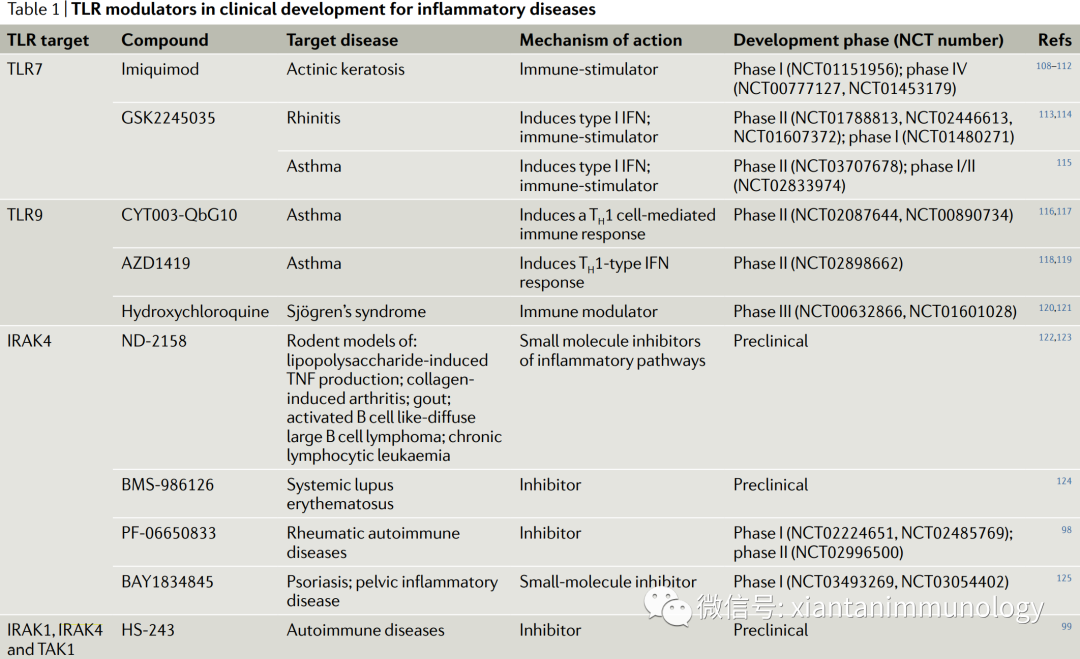
TLR7和TLR9的靶向分子,主要适应症是针对哮喘,鼻炎等炎症性疾病。IRAK4的小分子抑制剂则多为针对自身免疫性疾病。
包括辉瑞的PF-06650833,施贵宝的BMS-986126,拜尔的BAY1834845等。
喵评:TLR7和TLR9一个促进SLE,一个保护抑制SLE。TLR7参与B细胞发育及抗体分泌浆细胞的分化,但是药物开发,还没有针对自免开展。